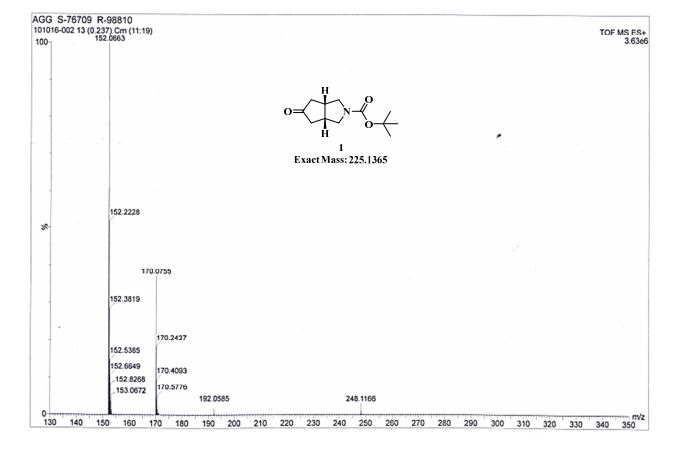
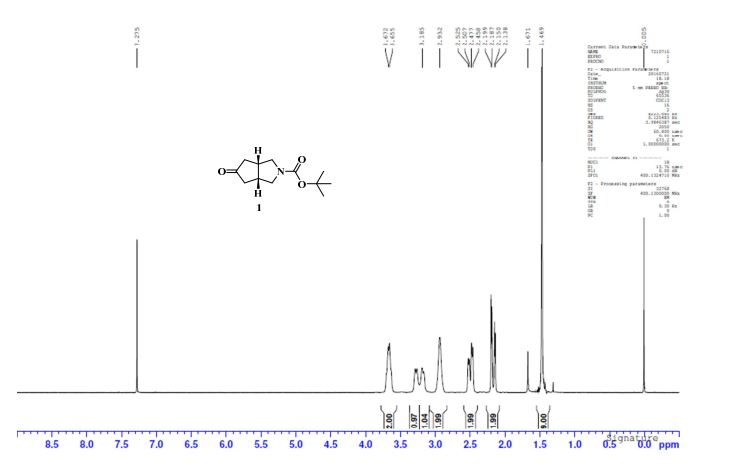
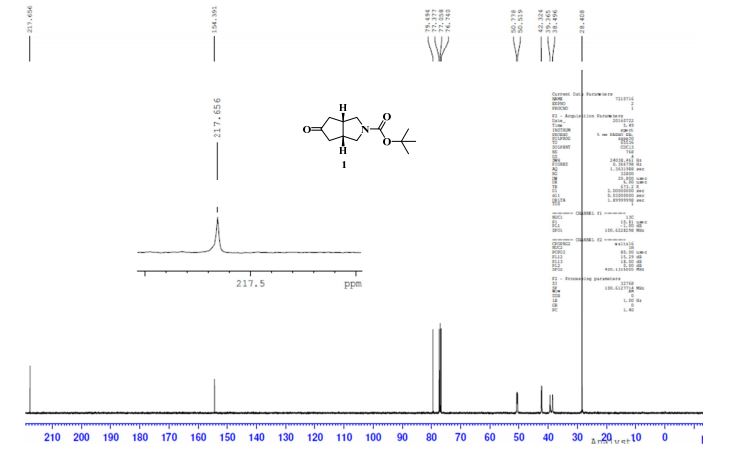
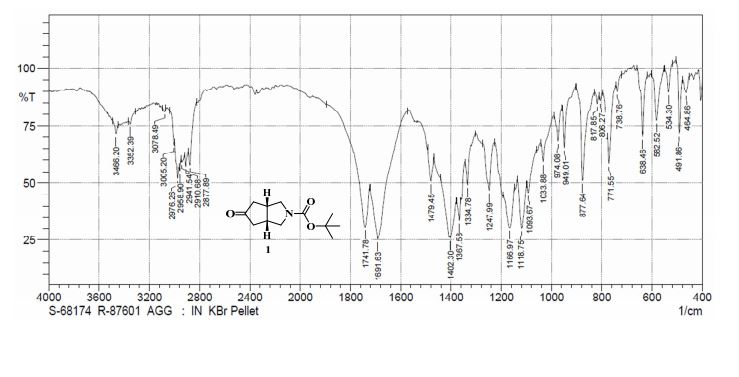
Example 2 - Preparation Of Compound 2 (l-(3,3-bis(3-(methylsulfonyI)phenyl)propyl)-4-(3- (methylsulfonyl) phenyl)piperidine)
Preparation of ethyl 3-(4-oxopiperidin-l-yl)-propanoate (starting material for Compound 2)
3-(4-oxopiperidin-1-yl)-propanoate
Ethanol (1550mL) was poured into a 4 L three-necked round-bottom flask equipped with over-head stirring followed by the addition of 125g (814mmol, leq) 4-piperidone monohydrate hydrochloride and 225g (1628mmol, 2eq) potassium carbonate. Ethyl 3-chloropropanoate (l l lg, leq) was added and the reaction mixture was stirred for 3h after which HPLC showed that the product reached only 10% area. Another 0.5eq of K2CO3 was added (56.2g) and stirring continued at 24°C. After total of 45h the product reached 86% area (HPLC). Another 0.2eq of K2CO3 was added and the reaction mixture was stirred for additional 4.5h at 35°C after which HPLC showed 96% area of the product. The mixture was filtered through a sintered glass filter, washed with 200 ml ethanol and concentrated on vacuum to 156g yellow colored oil that was distilled under vacuum of 2mmHg in 156°C bath. The main fraction distilled at 120°C to yield 97.8g (60%) of 99.3% area (HPLC).
Preparation of l -('3-hvdroxy-3,3-bis(3-(methylthio)phenyl')propyl)-4-(3-(methylthio) phenyl)piperidin-4-ol (Compound 2, 1st intermediate)
- - me y t o pheny p peri n-4-ol
3-Bromothioanisole (170.3g; 0.84mol, 3.2eq) and THF (700mL) were charged to a 2L flask, stirred under nitrogen and cooled on dry ice/acetone bath to -74°C. A solution of n-hexyllithium in hexane (2.3M; 237.4g; 0.77mol, 3.0eq) was added and the reaction mixture became slightly yellowish. Stirring continued for additional 30min at -74°C. A solution of ethyl 3-(4-oxopiperidin-l-yl)propanoate (50.2g; 0.26 mol, leq) in THF (lOOmL) was added during lhl5min to the reaction mixture and the stirring continued for additional 30min at -74°C to give a yellow clear solution. The cooling stopped and the reaction warmed to -40°C. A solution of HC1 (33%; 90g, 0.82mol, 3. leq) in water (lOOmL) was added dropwise for 20min to give a light yellow emulsion in +8°C. The light yellow organic phase was separated, washed with water (3x200mL) and extracted twice with aqueous HC1 (33%HC1 40g/300mL water) to give lower yellow phase (234g). The organic upper light yellow phase was evaporated up to 159g solution and the precipitate formed during concentration was filtered to give 19. lg yellow sticky precipitate. The precipitate was combined with the lower yellow phase, methanol (50mL) and THF (200mL) were added and distilled (67°C, 248g distilled). Heptane (200mL was added, the two liquid phase was stirred for 20min at 40°C and cooled to RT. The upper heptane phase was discarded and water (200mL) was added to the viscous yellow residue water. After stirring stopped the colorless water was decanted to leave 182g of very viscous light yellow residue (HPLC: 82% area).
Preparation of l-(3,3-bis(3-(methylthio)phenyl)allyl)-4-(3-(methylthio)phenyl)-l,2,3.6-tetrahydropyridine (Compound 2, 2nd intermediate')

Into the viscous light yellow residue was added 2-propanol (200mL) and the reaction mixture was distilled at atmospheric pressure to give 200mL of azeotropic distillate, leaving dark yellow oil into which methanol (50mL), 2-propanol (350mL) and cone, sulfuric acid (36.5g, 0.35mol. 1.35eq) were added. The reaction mixture was heated for 26 hours (mixture temperature 81-84°C, vapor temperature 79°C) and about 440mL of distillate were collected. At the end the temperature reached 87°C and the reaction mixture was foaming. After cooling was added toluene (lOOmL) and water (200mL) and the reaction mixture was heated to reflux (87°C). The heating stopped and after cooling three phases were formed. The lower oily phase was washed with water (2x200mL) and concentrated by vacuum distillation to give dark yellow viscous residue. Water (300mL) was added and the mixture was refluxed then cooled to 40°C and water phase was decanted to leave about 200g orange turbid liquid (HPLC: 82% area) which was used in the next step.
Preparation of l-(3.3-bis(3-(methylsulfonyl)phenyl')allyl)-4-(3-(methylsulfonyl) phenyl)- 1,2,3,6-tetrahydropyridine (Compound 2, 3rd intermediate)
To the 200g orange turbid liquid from the previous stage was added 500mL water, sodium tungstate dihydrate (2g, 6mmol) and concentrated sulfuric acid (20mL). The mixture was heated to 35°C and
33%¾θ2 was added drop-wise in lh during which the yellow viscous mass on the bottom of the flask dissolved slowly and the temperature rose up to 55°C then decreased slowly to 42°C. The reaction mixture was heated to 50°C for 2hr and additional 32g of 33%¾θ2 were added. The reaction continued for another 4h at 50°C and additional 20g of 33%¾θ2 were added. After 2h the reaction mixture was cooled down (25°C) and alkalized to pH12 by 50%NaOH solution. Water (300mL) was added and after 20min of mechanical stirring was discarded. Another 200mL of water were added, stirred mechanically for 20min and discarded to give 158.2g highly viscous yellow mass (HPLC: 75.4% area). This mass was heated for 30min 4 times with butanol (200mL@95°C, 200mL@ 100°C, 400mL@ 100°C and 700mL@ 114°C) and twice with acetic acid (8mL and 250mL@95°C) to give light brown oil that was used in the next step (114.9g, HPLC: 89% area).
Preparation of l-(3.3-bis(3-(methylsulfonyl)phenyl)propyl)-4-("3-(methylsulfonyl) phenvDpiperidine (Compound 2)

The light brown oil from the previous stage (114.9g, HPLC: 89% area) was added into a 2L autoclave with 550mL acetic acid and 10%Pd/C catalyst (25g, 23.5mmol). Hydrogen was introduced (120psi) and the reaction was heated to 90°C for 16h. After cooling, the catalyst was filtered, washed with acetic acid (50ml) and the clear yellowish filtrate was concentrated in vacuum to give 134g brown viscous residue (HPLC: 82% area). Water (300ml) was added, made alkaline (40% NaOH, pH>12) and extracted with 120mL dichloromethane that after concentration gave 77.2g brown sticky mass (HPLC: 83% area). The residue was treated with butanol (5xl00mL, 95°C), cooled down and the butanol phase over an oily phase was filtered. A total of 74.9g solid phase was resulted which was dissolved in 200mL acetone and the clear yellow solution was evaporated to give 70. lg dark yellow clear viscous residue. The residue was treated with heptane (2xl00mL, 95°C) which was cooled and decanted. After evaporation in a rotavapor a pale yellow foamy solid was obtained (65. lg, HPLC: 84% area). The solid was dissolved in 200mL dichloromethane, 85g silica was added and the mixture was evaporated and loaded on 1.32Kg silica gel column which was eluted by dichloromethane with 0.5-3.0% methanol and 0.5% triethylamine. Compound 2 was isolated to give 25.8g, HPLC: 93.2% area, lH-
NMR assay: 91.2%.
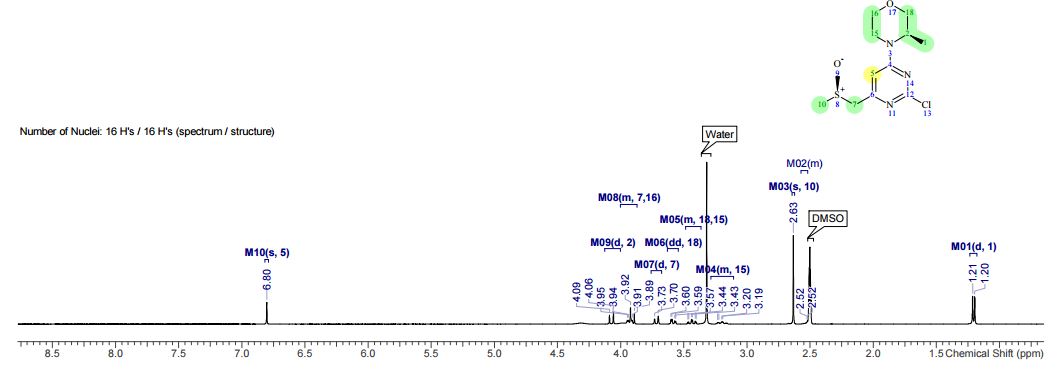
NMR Identity Analysis of Compound 2
Compound 2:

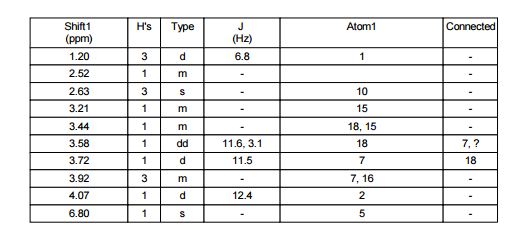
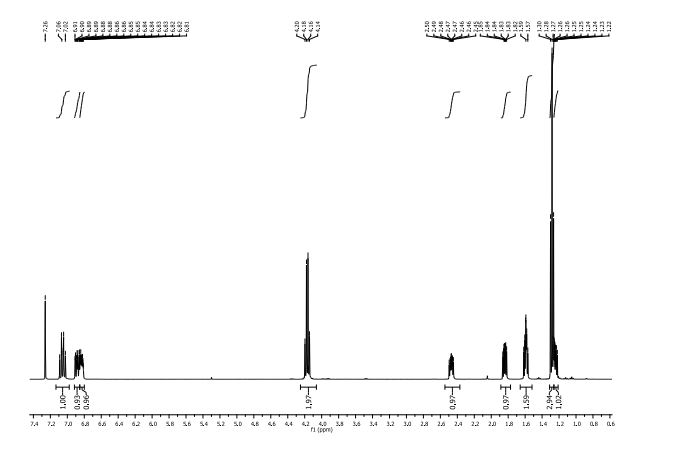
The following data in Tables 4 and 5 was determined using a sample of 62.03 mg Compound 2, a solvent of 0.6 ml CDC13, 99.8 atom%D, and the instrument was a Bruker Avance III 400 MHz.
Table 4: Assignment of ¾
NMRa-
c
a The assignment is based on the coupling pattern of the signals, coupling constants and chemical shifts.
b Weak signal.
c Spectra is calibrated by the solvent residual peak (7.28 ppm).
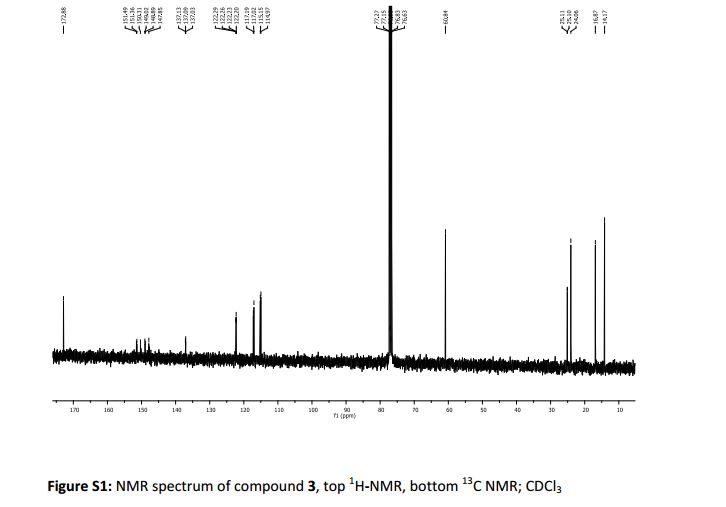
Table 5: Assignment of
13C
NMRa,b
a The assignment is based on the chemical shifts and 1H-13C couplings extracted from HSQC and HMBC experiments.
b Spectra is calibrated by a solvent peak (77.16 ppm)