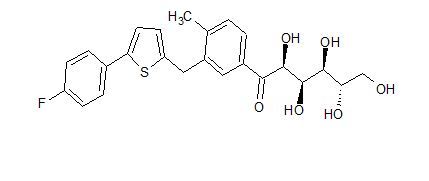
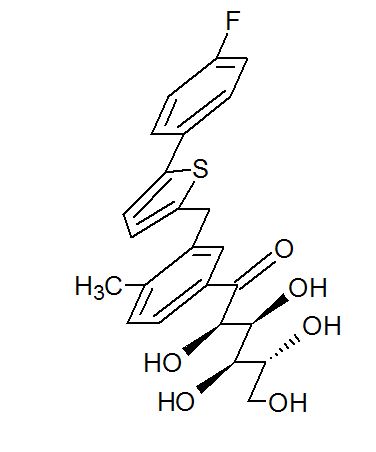
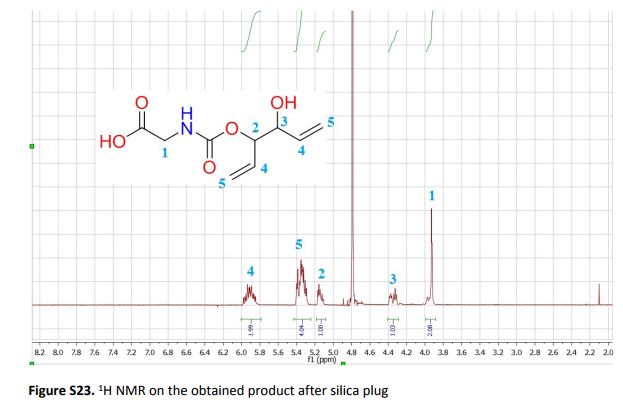
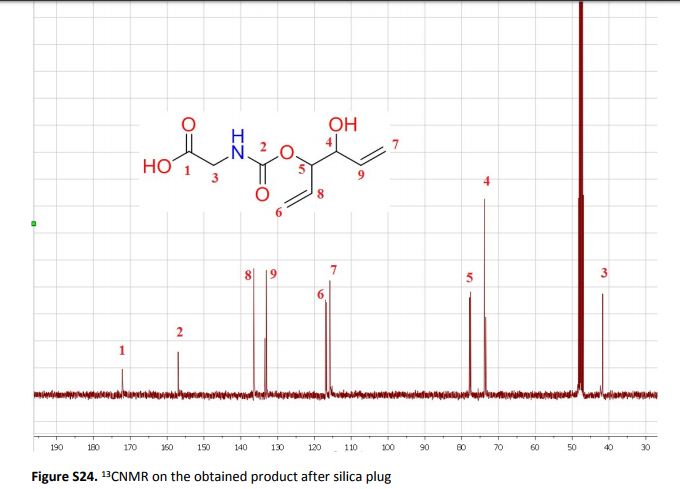
Green Chem., 2018, Advance Article
DOI: 10.1039/C7GC02627G, Paper
DOI: 10.1039/C7GC02627G, Paper
Kathiravan Murugesan, Thirusangumurugan Senthamarai, Manzar Sohail, Muhammad Sharif, Narayana V. Kalevaru, Rajenahally V. Jagadeesh
Nanoscale Fe2O3-catalyzed environmentally benign synthesis of nitriles and amides has been performed from easily accessible aldehydes and ammonia using O2.
Nanoscale Fe2O3-catalyzed environmentally benign synthesis of nitriles and amides has been performed from easily accessible aldehydes and ammonia using O2.
Stable and reusable nanoscale Fe2O3-catalyzed aerobic oxidation process for the selective synthesis of nitriles and primary amides
Abstract
The sustainable introduction of nitrogen moieties in the form of nitrile or amide groups in functionalized molecules is of fundamental interest because nitrogen-containing motifs are found in a large number of life science molecules, natural products and materials. Hence, the synthesis and functionalization of nitriles and amides from easily available starting materials using cost-effective catalysts and green reagents is highly desired. In this regard, herein we report the nanoscale iron oxide-catalyzed environmentally benign synthesis of nitriles and primary amides from aldehydes and aqueous ammonia in the presence of 1 bar O2 or air. Under mild reaction conditions, this iron-catalyzed aerobic oxidation process proceeds to synthesise functionalized and structurally diverse aromatic, aliphatic and heterocyclic nitriles. Additionally, applying this iron-based protocol, primary amides have also been prepared in a water medium.

cas 19102-07-9
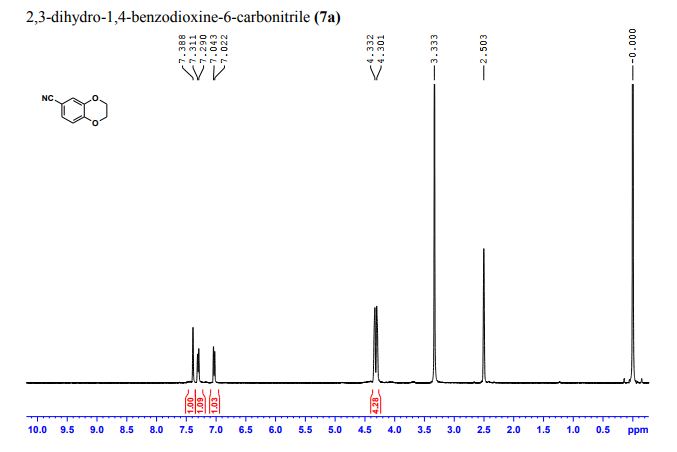
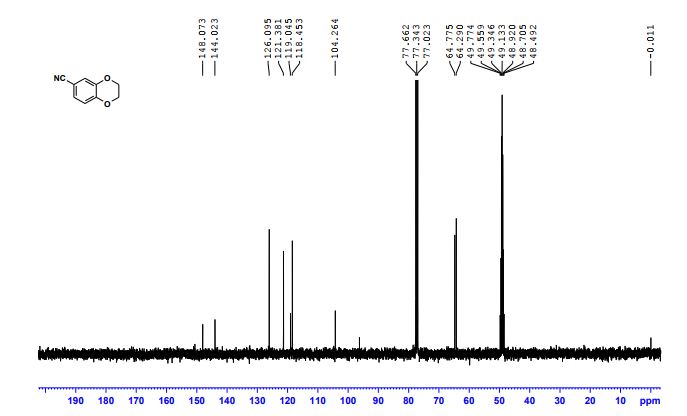
- 1,4-Benzodioxan-6-carbonitrile (8CI)
- 2,3-Dihydro-1,4-benzodioxin-6-carbonitrile
- 1-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)nitrile
MP
| Melting Point, °C | ||
|---|---|---|
| 105 - 106 Tetrahedron, 2015, vol. 71, 29, p. 4883 - 4887 |
NMR PREDICTS
1H NMR
13C NMR PREDICT
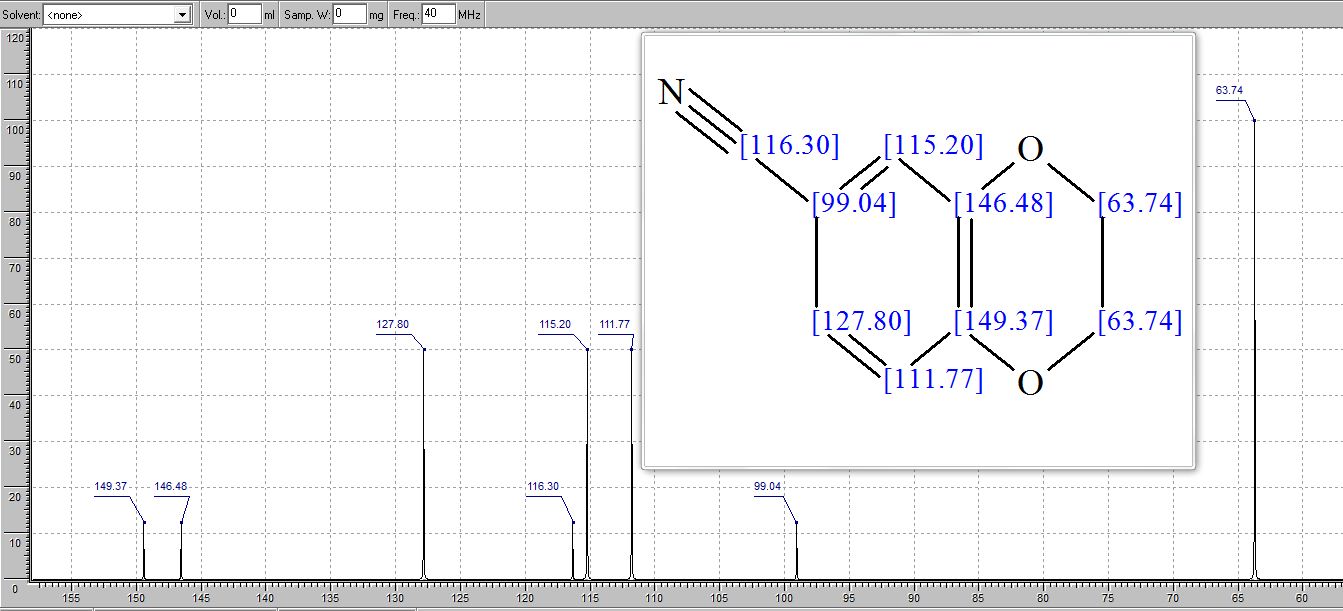
More................
Journal of the American Chemical Society, 2001, vol. 123, 49, p. 12202 - 12206

More.............
RSC Advances, 2013, vol. 3, 44, p. 22389 - 22396

MORE........
Organic Letters, 2017, vol. 19, 12, p. 3095 - 3098
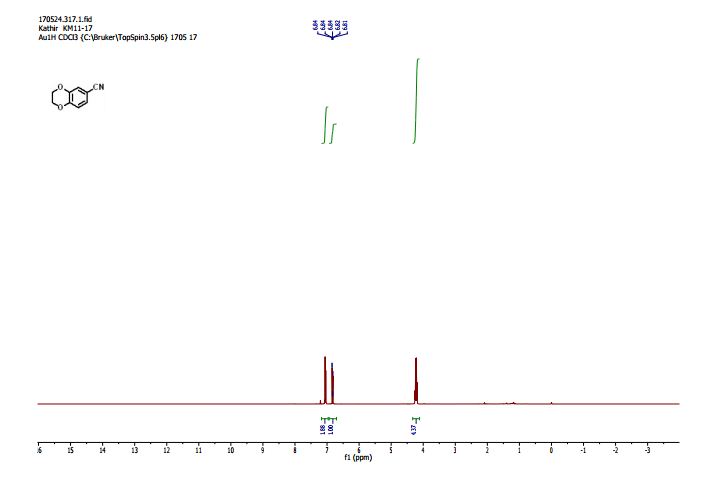
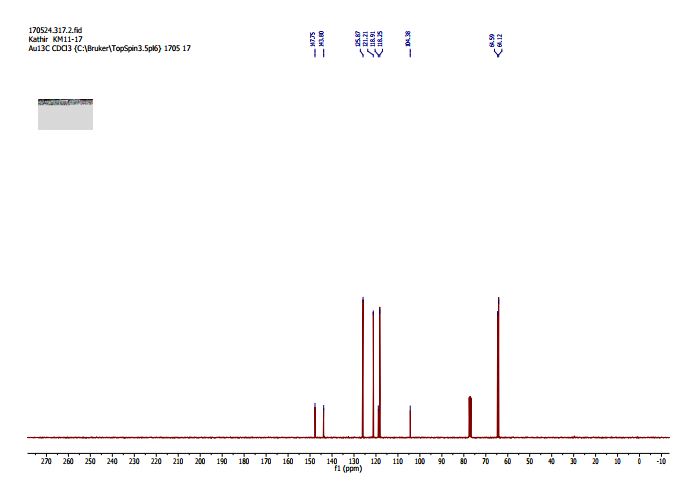
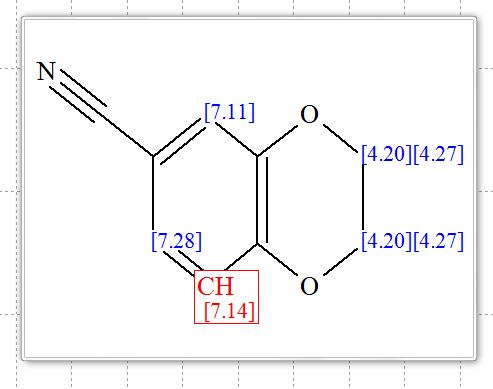
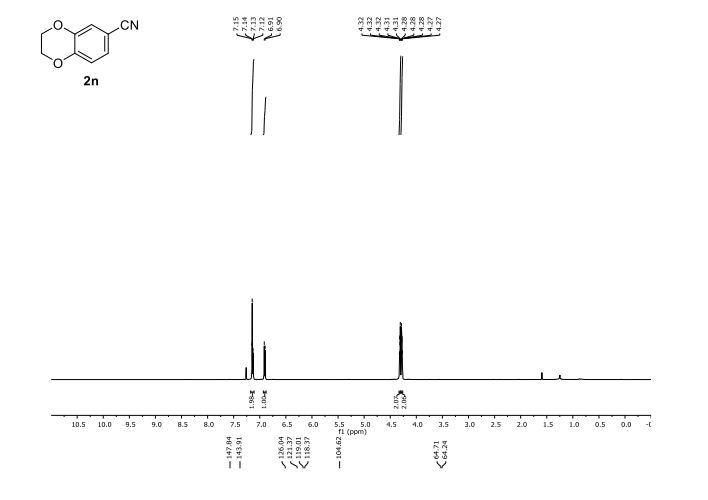
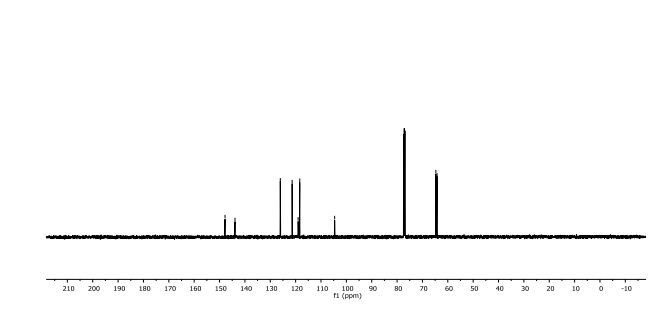
2,3-Dihydrobenzo[b][1,4]dioxine-6-carbonitrile (Scheme 1, 2n) According to the general procedure A, the reaction of 1n (0.20 mmol), zinc cyanide (2.0 equiv), PCyPh2 (0.20 equiv) and Pd(OAc)2 (0.05 equiv) in dioxane (0.25 M) for 16 h at 150 °C, afforded after work-up and chromatography the title compound in 75% yield (24.2 mg). White solid. 1H NMR (500 MHz, CDCl3) δ 7.17-7.11 (m, 2H), 6.91 (d, J = 8.1 Hz, 1H), 4.32-4.31 (m, 2H), 4.30- 4.26 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 147.84, 143.91, 126.04, 121.37, 119.01, 118.37, 104.62, 64.71, 64.24.
//////////////