
Tuesday, 25 November 2014
HMBC (multiple-bond CH correlation) of codeine
HMBC (multiple-bond CH correlation) of codeine
This is a 2D experiment used to correlate, or connect, 1H and 13C peaks for atoms separated by multiple bonds (usually 2 or 3). The coordinates of each peak seen in the contour plot are the 1H and 13C chemical shifts. This is extremely useful for making assignments and mapping out covalent structure.
The information obtained is an extension of that obtained from an HMQC spectrum, but is more complicated to analyze. Like HMQC, this is an "inverse detection" experiment, and is possible only on newer model spectrometers. Acorn NMR's new JEOL Eclipse+ 400 is equipped to perform inverse experiments, and uses Z-gradients for improved spectral quality.
The time required for an HMBC depends on the amount of material, but is much greater than for HMQC, and can take from an hour to overnight.
The contour plot shown below is of 3.3 mg codeine in ~ .65 ml CDCl3. See also comparison to the HMBC spectrum of an 18 mg sample.
Normal 1D 1H and 13C spectra are shown along the edges. Peaks occur at coordinates in the 2 dimensions corresponding to the chemical shifts of a carbon and protons separated by (usually) 2 or 3 bonds. The experiment is optimized for couplings of ~8 Hz. Smaller couplings are observed, but their intensities are reduced. Compare to the spectrum obtained when the experiment is optimized for 4 Hz.
The experiment is designed to suppress 1-bond correlations, but a few are observed in most spectra. In concentrated samples of conjugated systems, 4-bond correlations can be observed. There is no way to know how many bonds separate an H and C when a peak is observed, so analysis is a process of attempting to assign all observed peaks, testing for consistency and checking to be sure none of the assignments would require implausible or impossible couplings.
Because of the large number of peaks observed, analysis requires several expanded plots. In this case, the spectrum has been divided into 4 sections, each of which is discussed below.

| The discussion below uses the numbering system shown at right. The numbers were assigned to peaks in the 1D 13C spectrum, starting downfield, moving upfield, and numbering each sequentially. This generates a unique identifier for each Carbon, even before knowing any assignments. |  |

In aromatic rings, the most common correlations seen in HMBC spectra are 3-bond correlations because they are typically 7-8 Hz, which is the value for which the experiment is optimized. The coupling constant is affected by substituents, so 2-bond correlations are also sometimes observed.
The red lines in the plot above show correlations from aromatic proton H-8 to aromatic carbons C-1 and C-6 (both are 3-bond couplings) and a weak correlation to C-2, a 2-bond coupling.
The other aromatic proton, H-7, has correlations to C-2 and C-4, both of which are 3-bond couplings.
The green lines in the plot above show correlations from proton H-9 to carbons C-1, C-3 and C-4 (all are 3-bound couplings). With the poor digital resolution of the spectrum in the carbon dimension (512 data points spread over 17kHz), the peaks for C-3 and C-4 run together because they are barely resolved.

The peaks indicated in red above are due to 1-bond coupling in CHCl3 solvent. Note that the pair of peaks don't line up with any H peaks, but are symmetrically located about the CHCl3 peak, with a separation equal to the 1-bond C-H coupling constant.
The other 2 peaks in this plot are H-7 to C-18 and H-9 to C-17.

The region in the above plot shows correlations between aliphatic Hs and aromatic Carbons. In the lower left corner is a peak showing the 3-bond coupling between the methoxy Hs and C-2, the aromatic carbon bearing the methoxy.
Two protons show correlations to the same 3 carbons. These are the geminal H-18 protons, showing coupling to C-7 and C-4 (both 3-bond couplings) and C-6 (2-bond coupling). The remaining peak is H-17 to C-4. Note that the corresponding coupling from H-17' is not observed. As with H-H couplings, the value of the 3-bond coupling constant is dependent on the dihedral angle.

The last quadrant is shown above, showing correlations between aliphatic Hs and Cs. The peaks are identified below, for each H, starting from the left. Carbons 14 and 15 are only 0.1 ppm apart, and are not resolved in the 2D spectrum.
H-11 to C-14 and/or C-15
H-18 to C-16, C-11
H-13 to C-17, C-14 and/or C-15, C-11
H-14 to C-13 and C-11
H-18' to C-11
H-17 to C14 and/or C-15, C-13
During the process of assigning HMBC peaks, it can be useful to indicate on the plot the positions of the 1-bond correlations. NUTS has the ability to do this, using the compare command. In the HMBC plot below, 1-bond HMQC peaks are indicated by X.

Acquisition Parameters:
512 complex points in direct dimension
128 t1 increments
8 scans
2 sec. relaxation delay
Total acquisition time: 35 min
Processing:
sine squared window function in both dimensions with 0 degree phase shift in t2 and 90 degree phase shift in t1
2x zero-fill in the indirect dimension
magnitude calculation (no phasing is required)
final data size 512 x 512
HMBC of codeine optimized for small couplings
See explanation of the HMBC experiment.
Long range C-H couplings cover a range of values, typically less than 10 Hz. The experiment is optimized for a specific coupling, and selection of that value is a trade-off. Correlations due to couplings of other values are reduced in intensity. To optimize for a specific J value, a delay in the pulse sequence is set to 1/(2J). For very small couplings, this delay becomes so long that much of the magnetization is lost to relaxation. Typically, the delay is optimized for J of 7-8 Hz, the expected value for aromatic 3-bond couplings. However, longer-range couplings can be observed if the delay is optimized for smaller couplings.
The contour plots shown below are of 8 mg codeine in ~ .65 ml CDCl3, in which the experiment was optimized for couplings of 4 Hz.
Compare to the HMBC spectrum optimized for 8 Hz.
Because of the large number of peaks observed, analysis requires several expanded plots. In this case, the spectrum has been divided into 4 sections, each of which is discussed below.

| The discussion below uses the numbering system shown at right. The numbers were assigned to peaks in the 1D 13C spectrum, starting downfield, moving upfield, and numbering each sequentially. This generates a unique identifier for each Carbon, even before knowing any assignments. | |

Compare to the same quadrant in the HMBC optimized for 8 Hz. Several additional 2-bond couplings are observed, as are 1-bond couplings for 7 and 8. The peaks indicated by red circles are 4-bond couplings: H-8 to C-4 and H-7 to C-1.

Compared to the 8 Hz-optimized spectrum, one additional peak is observed in this quadrant, indicated by the red circle. This is a 4-bond coupling from H-7 to C-15.

In this quadrant, three 4-bond peaks are indicated by red circles (H-18 to C-8 and C-1; H-18' to C-8). In addition, two 5-bond couplings are indicated by blue circles. These are H-18 and H-18' to C-2.

The only additional correlations in the quadrant not seen in the 8 Hz-optimized spectrum are from H-13' (just upfield of the intense N-Me singlet) to carbons 17, 14/15 and 11.
Acquisition Parameters:
512 complex points in direct dimension
128 t1 increments
64 scans
2 sec. relaxation delay
Total acquisition time: 4.5 hrs
Processing:
sine squared window function in both dimensions with 0 degree phase shift in t2 and 90 degree phase shift in t1
2x zero-fill in the indirect dimension
magnitude calculation (no phasing is required)
final data size 512 x 512
HMQC (1-bond CH correlation) of codeine
HMQC (1-bond CH correlation) of codeine
This is a 2D experiment used to correlate, or connect, 1H and 13C peaks for directly bonded C-H pairs. The coordinates of each peak seen in the contour plot are the 1H and 13C chemical shifts. This is helpful in making assignments by comparing 1H and 13C spectra.
This experiment yields the same information as the older "HETCOR" experiment, but is more sensitive, so can be done in less time and/or with less material. This is possible because in the HMQC experiment, the signal is detected by observing protons, rather than carbons, which is inherently more sensitive, and the relaxation time is shorter. This so-called "inverse detection" experiment is technically more difficult and is possible only on newer model spectrometers. Acorn NMR's new JEOL Eclipse+ 400 is equipped to perform inverse experiments, and uses Z-gradients for improved spectral quality.
The time required for an HMQC depends on the amount of material, but can be done in 1/2 hour or less, compared to several hours for a HETCOR spectrum.

Contour plot of the HMQC spectrum. Because it is a heteronuclear experiment, the 2 axes are different, and the plot is not symmetrical. Unlike a COSY spectrum, there are no diagonal peaks.
Normal 1D 1H and 13C spectra are shown along the edges. Peaks occur at coordinates in the 2 dimensions corresponding to the chemical shifts of a carbon and its directly bonded proton(s). For example, the contour peak indicated in red shows that the 13C with peak at 91.5 ppm is bonded to the 1H with peak at 4.9 ppm.
Non-equivalent methylene protons are easily identified as 2 peaks located at the same 13C position. There are 3 CH2s in the codeine HMQC spectrum.
| |
The sample is 3.3 mg codeine in ~ .65 ml CDCl3
Acquisition Parameters:
512 complex points in direct dimension
128 t1 increments
2 scans
2 sec. relaxation delay
Total acquisition time: ~ 10 min.
Processing:
sine squared window function in both dimensions with 45 degree phase shift
2x zero-fill in the indirect dimension
magnitude calculation (no phasing is required)
final data size 512 x 512
There are variations on this experiment, including a version in which CH2s have phase opposite of that of CH and CH3 peaks, called an HSQC-DEPT spectrum. Negative peaks are shown in red in the plot below, easily identifying the 3 CH2s in codeine.

Synthesis, characterization, and photophysical properties of a thiophene-functionalized bis(pyrazolyl) pyridine (BPP) tricarbonyl rhenium(I) complex

Dalton Trans., 2010,39, 7692-7699
DOI: 10.1039/C0DT00357C
A bromo tricarbonyl rhenium(I) complex with a thiophene-functionalized bis(pyrazolyl) pyridineligand (L), ReBr(L)(CO)3 (1), has been synthesized and characterized by variable temperature and COSY 2-D 1H NMR spectroscopy, single-crystal X-ray diffraction, and photophysical methods. Complex 1 is highly luminescent in both solution and solid-state, consistent with phosphorescence from an emissive 3MLCT excited state with an additional contribution from aLC 3(π→π*) transition. The single-crystal X-ray diffraction structure of the title ligand is also reported.

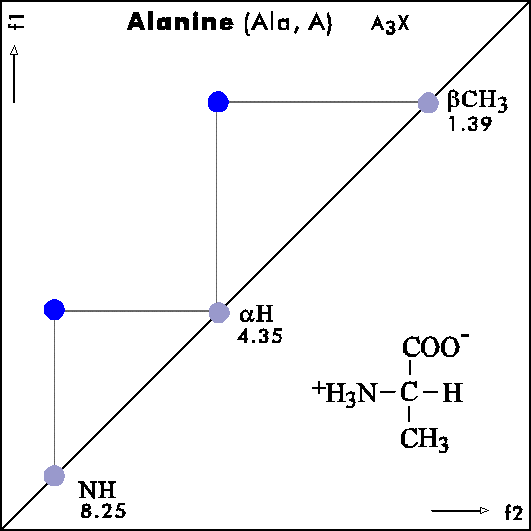
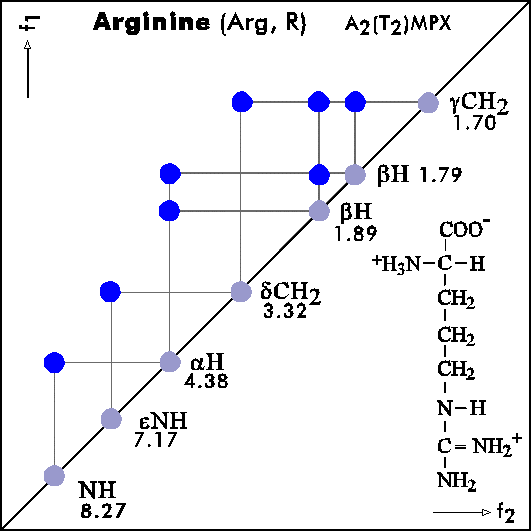
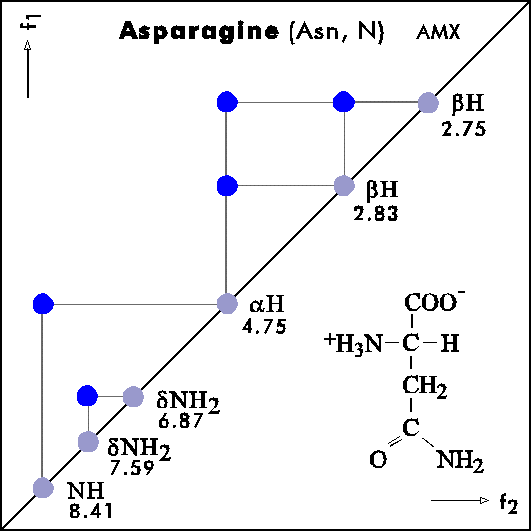
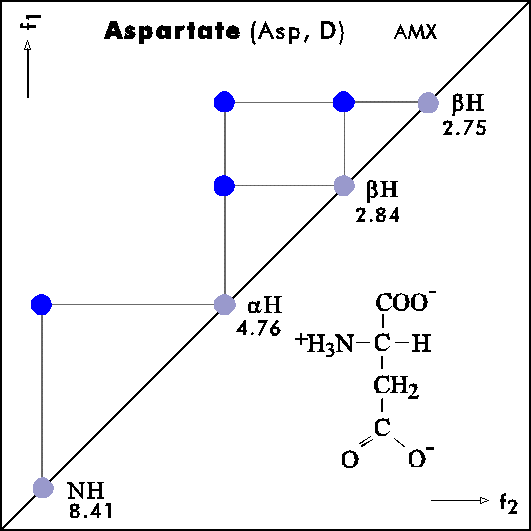
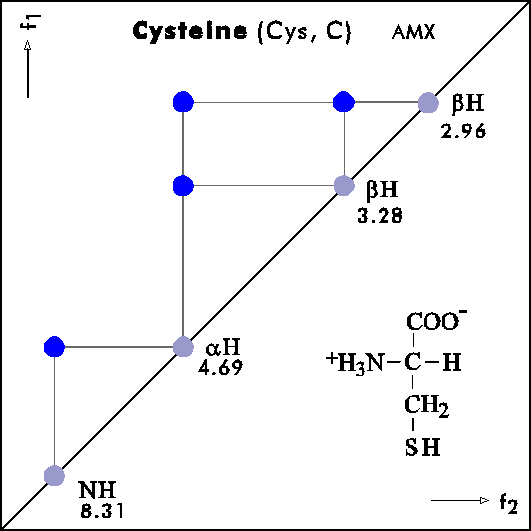
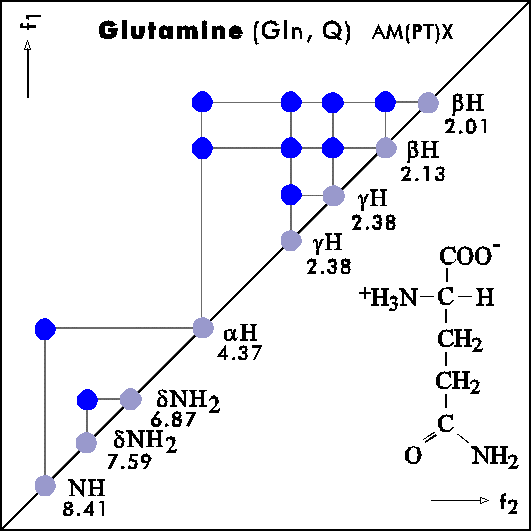
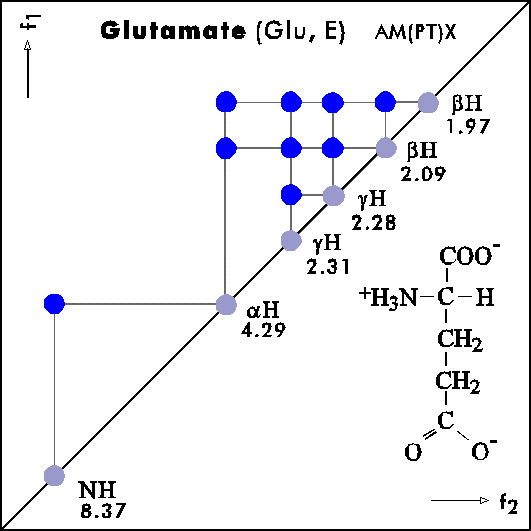
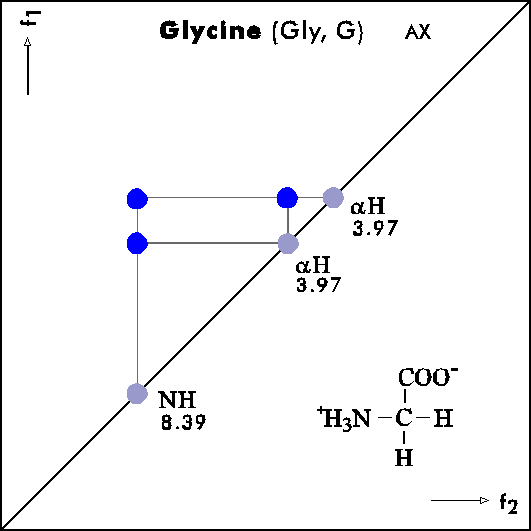
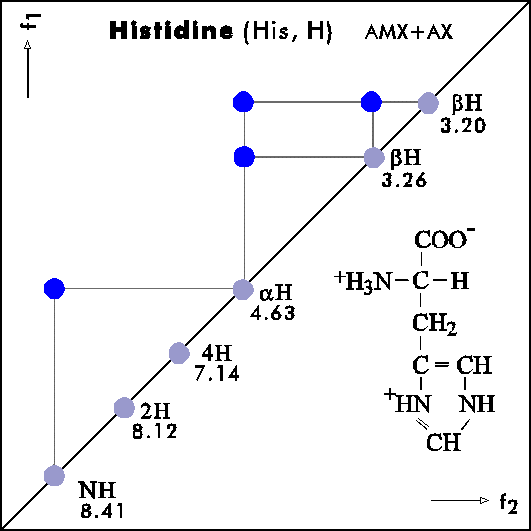
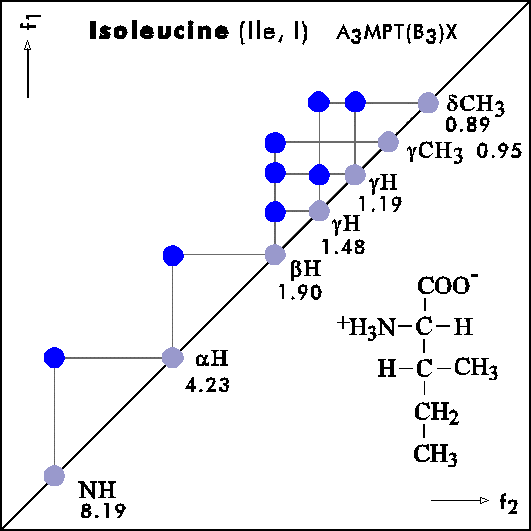
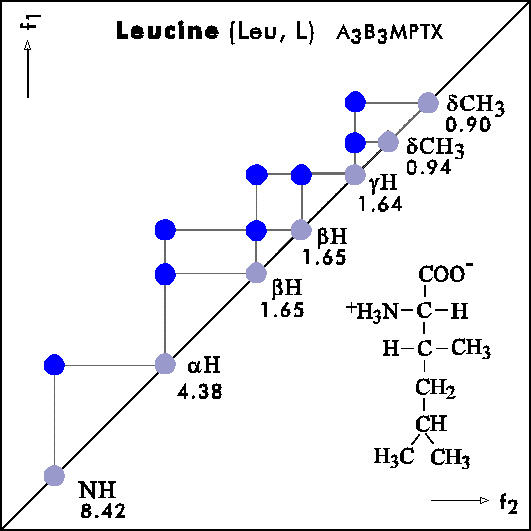
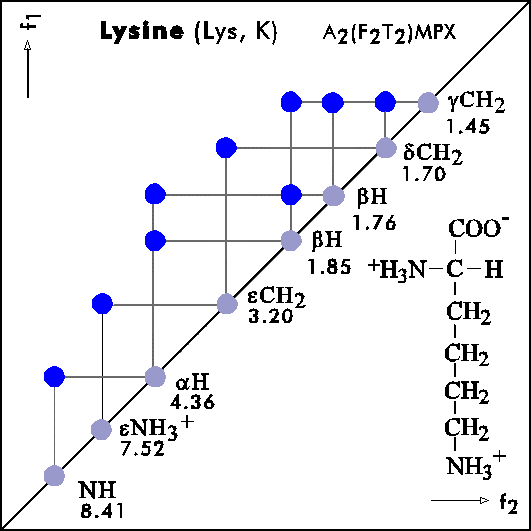
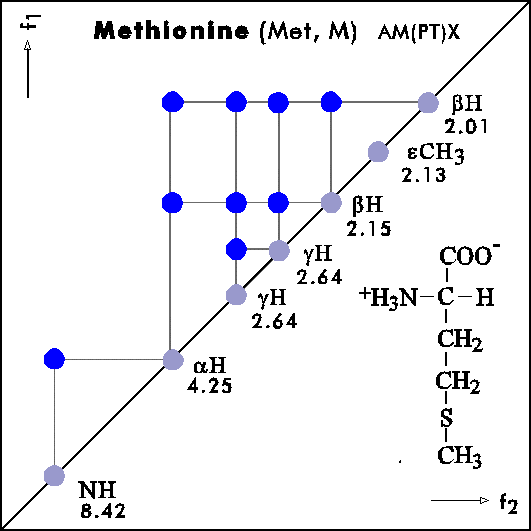
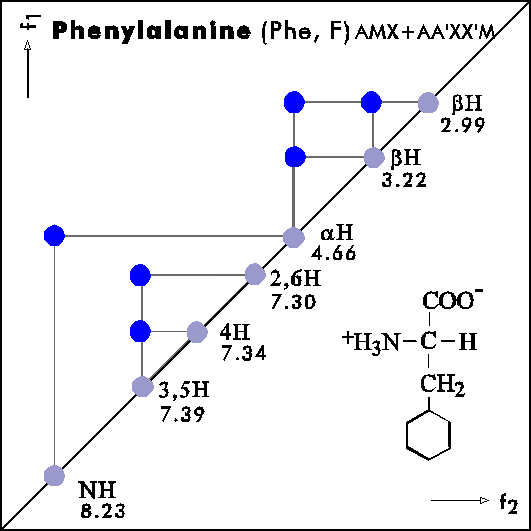
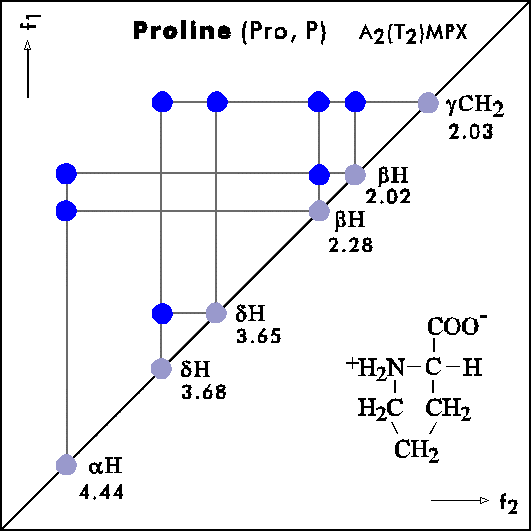
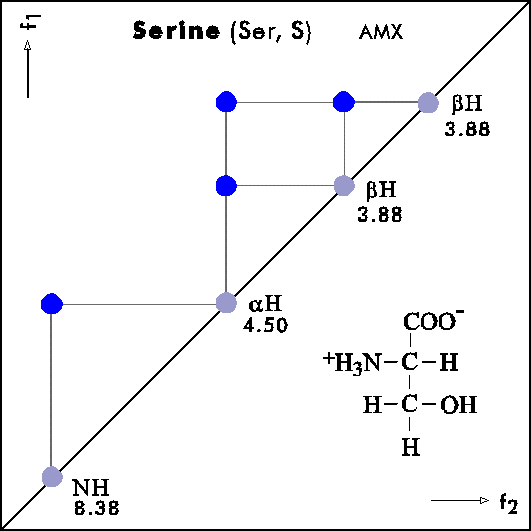
COSY PATTERNS OF AMINO ACIDS

.................
Cosy-Pattern of Amino Acids
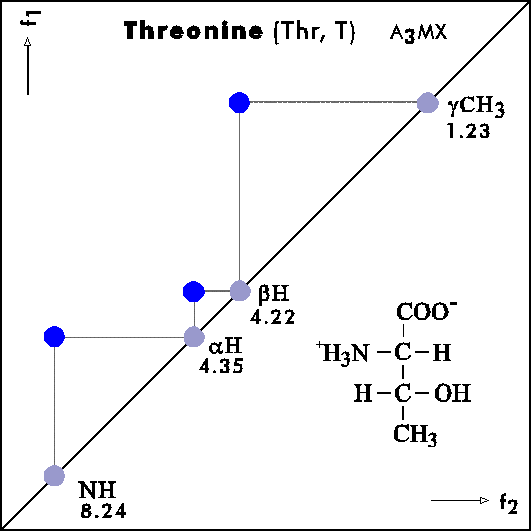
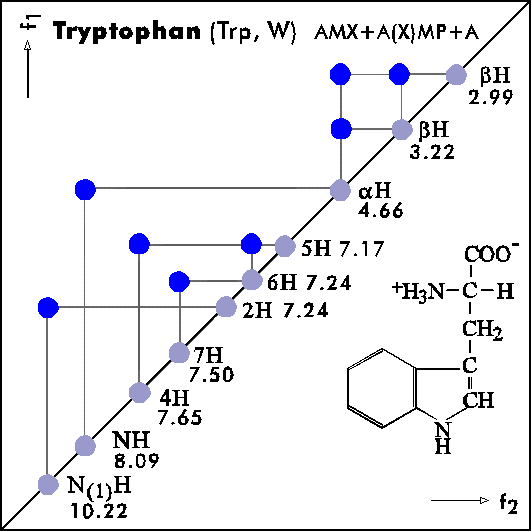
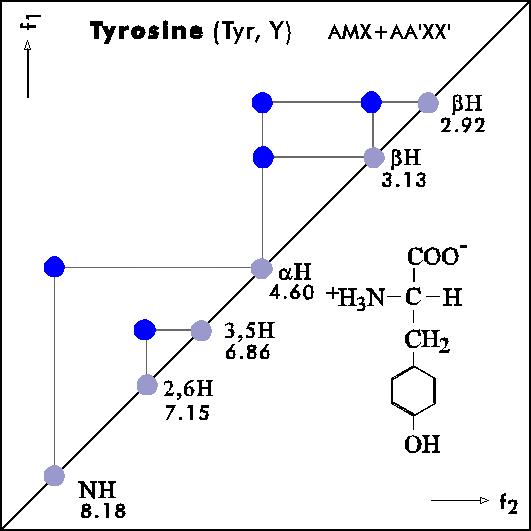
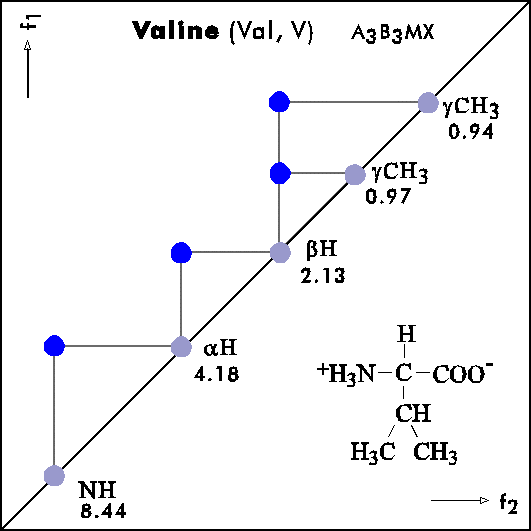
Name Abbr. Linear structure formula ====================================================== Alanine Ala A CH3-CH(NH2)-COOH Arginine Arg R HN=C(NH2)-NH-(CH2)3-CH(NH2)-COOH Asparagine Asn N H2N-CO-CH2-CH(NH2)-COOH Aspartic acid Asp D HOOC-CH2-CH(NH2)-COOH Cysteine Cys C HS-CH2-CH(NH2)-COOH Glutamine Gln Q H2N-CO-(CH2)2-CH(NH2)-COOH Glutamic acid Glu E HOOC-(CH2)2-CH(NH2)-COOH Glycine Gly G NH2-CH2-COOH Histidine His H N=C-NH-C=C-CH2-CH(NH2)-COOH |________| Isoleucine Ile I CH3-CH2-CH(CH3)-CH(NH2)-COOH Leucine Leu L (CH3)2-CH-CH2-CH(NH2)-COOH Lysine Lys K H2N-(CH2)4-CH(NH2)-COOH Methionine Met M CH3-S-(CH2)2-CH(NH2)-COOH Phenylalanine Phe F Ph-CH2-CH(NH2)-COOH Proline Pro P NH-(CH2)3-CH-COOH |_________| Serine Ser S HO-CH2-CH(NH2)-COOH Threonine Thr T CH3-CH(OH)-CH(NH2)-COOH Tryptophan Trp W Ph-NH-CH=C-CH2-CH(NH2)-COOH |_______| Tyrosine Tyr Y HO-p-Ph-CH2-CH(NH2)-COOH Valine Val V CH3-CH(CH2)-CH(NH2)-COOH
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
All together now
COSY VS DOUBLER QUANTUM FILTERED COSY
COSY spectra are very useful in structure elucidation as they provide correlations between coupled spins. Often, NMR spectra have large singlet signals from uncoupled protons (such as t-butyl methyls, methoxy protons, excess water or a solvent signal) which provide no information in the COSY spectrum and perhaps even get in the way of looking for smaller coupled spins. In such cases one can use a double quantum filtered COSY sequence rather than a standard COSY 90 or COSY 45 sequence. Double quantum filtered COSY spectra filter out uncoupled singlets. A comparison of a standard COSY 90 and a double quantum filtered COSY sequence for ethyl acetate is shown below. One can see that the singlet is present in the COSY 90 spectrum but absent in the double quantum filtered COSY

HMQC

Only directly bonded hydrogen and carbons will give cross peaks (quaternary carbons are not seen), which makes interpretation rather straight foreword. As seen in the simulated spectrum below, assignment is made by drawing two lines at a right angle from the 1H spectrum to the 13C spectrum through the cross-peak, which looks like a series of concentric ellipses. Thus, in the spectrum below, C1 is directly attached to H1.
Subscribe to:
Comments (Atom)