Continuous niobium phosphate catalysed Skraup reaction for quinoline synthesis from solketal
Green Chem., 2017, Advance Article
DOI: 10.1039/C6GC03140D, Paper
Jing Jin, Sandro Guidi, Zahra Abada, Zacharias Amara, Maurizio Selva, Michael W. George, Martyn Poliakoff
Solketal is derived from the reaction of acetone with glycerol, a by-product of the biodiesel industry. We demonstrate the use of NbOPO4 as a catalyst for the conversion of solketal and anilines to quinolines
Green Chem., 2017, Advance Article
DOI: 10.1039/C6GC03140D, Paper
Jing Jin, Sandro Guidi, Zahra Abada, Zacharias Amara, Maurizio Selva, Michael W. George, Martyn Poliakoff
Solketal is derived from the reaction of acetone with glycerol, a by-product of the biodiesel industry. We demonstrate the use of NbOPO4 as a catalyst for the conversion of solketal and anilines to quinolines





Synthesis of 4-(quinolin-6-yl methyl)aniline (6a)
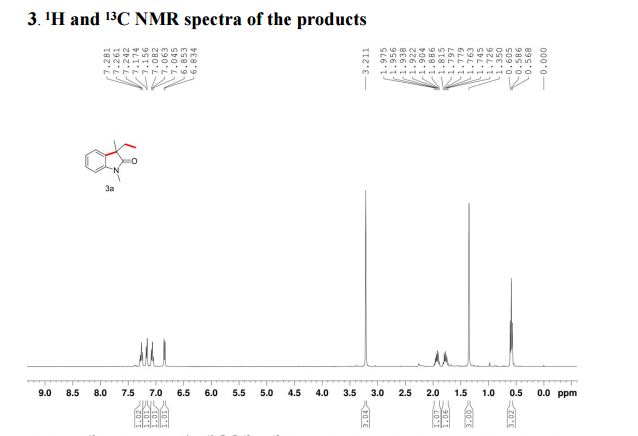
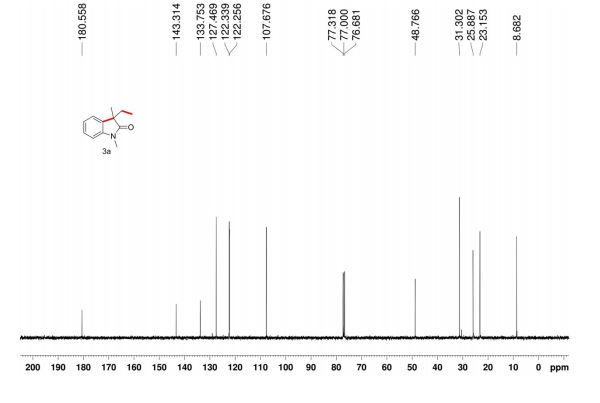
The reaction was carried out accordingly to the general procedure. The purification of 4-(quinoline-6-yl methyl)aniline 6a was carried out with a gradient of polarity from 80:20 to 30:70 (v/v) of CyHex:AcOEt as eluent. 1H NMR (400 MHz, CDCl3) δ ppm: 8.85 (dd, J=4.3,1.7Hz, 1H), 8.07 (dd, J=8.3,1.8Hz, 1H), 8.01 (d, J=9.2Hz, 1H), 7.58–7.54 (m, 2H), 7.36 (dd, J=8.3,4.2Hz, 1H), 7.02 (d, J=8.3Hz, 2H), 6.67–6.63 (m, 2H), 4.06 (s, 2H). 13C NMR (100 MHz, CDCl3) δ ppm: 149.9, 147.3, 144.9, 140.7, 135.9, 131.4, 130.6, 130.1, 129.5, 128.5, 126.6, 121.2, 115.5, 41.2. HRMS-ESI for C16H15N2 [M+H]+ calculated 235.1235, found 235.1245.
Continuous niobium phosphate catalysed Skraup reaction for quinoline synthesis from solketal
Abstract
Solketal is derived from the reaction of acetone with glycerol, a by-product of the biodiesel industry. We report here the continuous reaction of solketal with anilines over a solid acid niobium phosphate (NbP), for the continuous generation of quinolines in the well-established Skraup reaction. This study shows that NbP can catalyse all the stages of this multistep reaction at 250 °C and 10 MPa pressure, with a selectivity for quinoline of up to 60%. We found that the catalyst eventually deactivates, most probably via a combination of coking and reduction processes but nevertheless we show the promise of this approach. We demonstrate here the application of our approach to synthesize both mono- and bis-quinolines from the commodity chemical, 4,4′-methylenedianiline.























 Open Access
Open Access