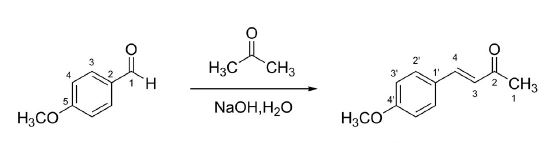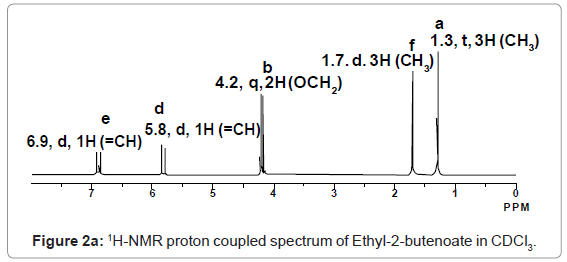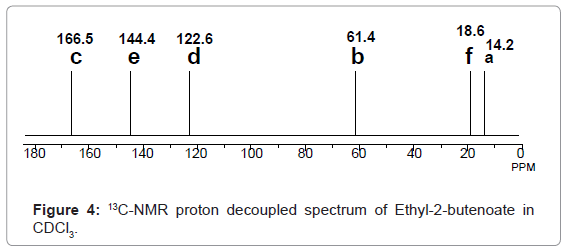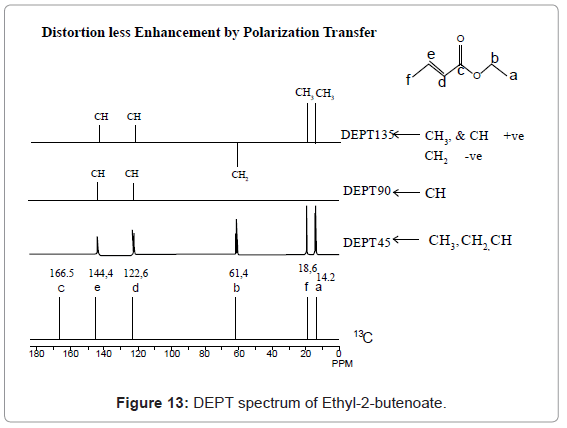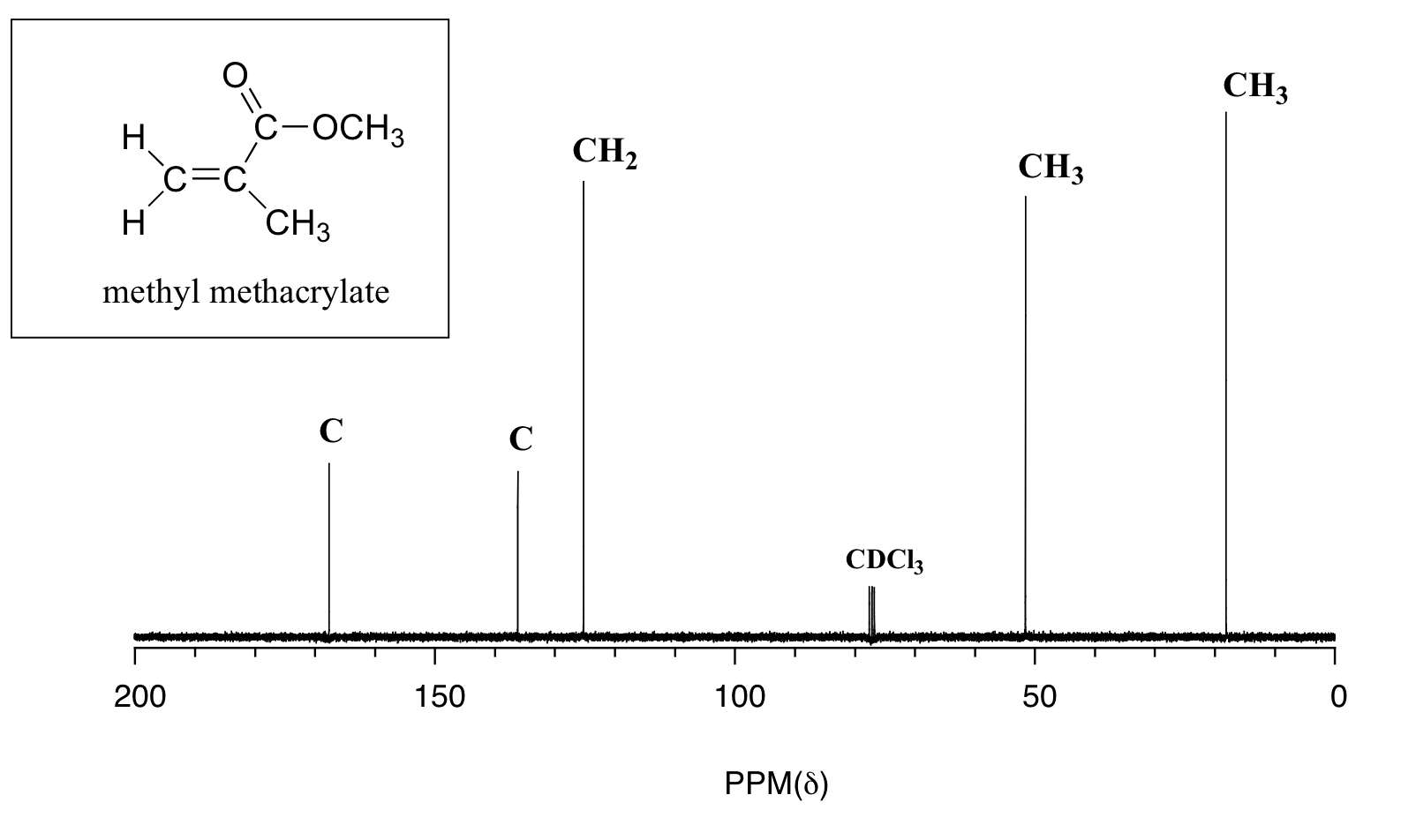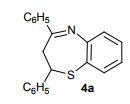
2,4-Diphenyl-2,3-dihydro-1,5-benzothiazepine (4a) 1,3-Diphenyl-2-propen-1-one (1a).
Yellow solid; mp 114-116 C [lit.1 , 114-115 °C], AcOEt/PE 1:9.
1H NMR (300 MHz, CDCl3,): 3.07 (t, J = 12.6 Hz, 1 H), 3.32 (dd, J = 4.7, 13.1 Hz, 1 H), 4.99 (dd, J = 4.5, 12.0 Hz, 1 H), 7.12-7.17 (m, 1 H), 7.25-7.30 (m, 5 H), 7.44-7.51 (m, 4 H), 7.62 (d, J = 6.1 Hz, 2 H), 8.06 (d, J = 7.5 Hz, 2 H).
Isolated Yield: 339 mg, 86%
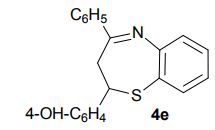
2-(4-Hydroxyphenyl)-4-phenyl-2,3-dihydro-1,5-benzothiazepine (4e) 3-(4-Hydroxyphenyl)-1-phenyl-2-propen-1-one (1e).
Light brown solid; mp 131-134 °C. AcOEt/PE 40:60.
1H NMR (CDCl3, 300 MHz): = 3.01 (t, J = 12.7 Hz, 1 H), 3.28 (dd, J = 4.8, 12.9 Hz, 1 H), 4.95 (dd, J = 4.7, 12.5 Hz, 1 H), 5.10 (bs, 1 H), 6.76 (d, J = 8.5 Hz, 2 H), 7.18-7.21 (m, 3 H), 7.35 (d, J = 8.5 Hz, 1 H), 7.46- 7.55 (m, 4 H), 7.63 (dd, J =1.5, 7.7 Hz, 1 H), 8.06 (m, 2 H).
13C NMR (CDCl3, 75 MHz): 37.99 (CH2), 60.07 (CH), 115.53 (CH), 123.08 (C), 127.40 (CH), 128.79 (CH), 131.17 (CH), 136.54 (C), 141.59 (C), 155.24 (C). IR (KBr): 1599, 2921, 3350 cm-1 .
MS (ESI): m/z= 332.24 (MH)+ . Anal. Calcd. for C21H17NOS: C, 76.10; H, 5.17; N, 4.23, found: C, 76.21; H, 5.15; N, 4.24. Isolated Yield: 360 mg, 87%


A practical synthesis of 2,3-dihydro-1,5-benzothiazepines
Green Chem., 2017, Accepted Manuscript
DOI: 10.1039/C7GC02097J, Paper
DOI: 10.1039/C7GC02097J, Paper
Domenico Carlo Maria Albanese, nicoletta gaggero, Meng Fei
2,3-Dihydro-1,5-benzothiazepines have been obtained through a domino process involving a Michael addition of 2-aminothiophenols to chalcones, followed by in situ cyclization. Up to 98% chemical yields have been obtained at...
2,3-Dihydro-1,5-benzothiazepines have been obtained through a domino process involving a Michael addition of 2-aminothiophenols to chalcones, followed by in situ cyclization. Up to 98% chemical yields have been obtained at...
A practical synthesis of 2,3-dihydro-1,5-benzothiazepines
Abstract
2,3-Dihydro-1,5-benzothiazepines have been obtained through a domino process involving a Michael addition of 2-aminothiophenols to chalcones, followed by in situ cyclization. Up to 98% chemical yields have been obtained at room temperature under essentially neutral conditions by using hexafluoro-2-propanol as an efficient medium.
/////////////////