Abstract
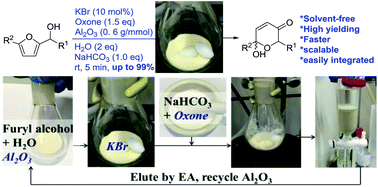
Reported here is the development of an environmentally friendly catalytic (KBr/oxone) and solvent-free protocol for the Achmatowicz rearrangement (AchR). Different from all previous methods is that the use of chromatographic alumina (Al2O3) allows AchR to proceed smoothly in the absence of any organic solvent and therefore considerably facilitates the subsequent workup and purification with minimal environmental impacts. Importantly, this protocol allows for scaling up (from milligram to gram), recycling of the Al2O3, and integrating with other reactions in a one-pot sequential manner.
A solvent-free catalytic protocol for the
Achmatowicz rearrangement
Author affiliations
*Corresponding authors
aDepartment of Chemistry, The Hong Kong University of Science and Technology, Clearwater Bay, Kowloon, China
E-mail: rtong@ust.hkFax: +(852)23581594
Tel: +(852) 23587357
bHKUST Shenzhen Research Institute, Shenzhen 518057, China
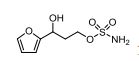
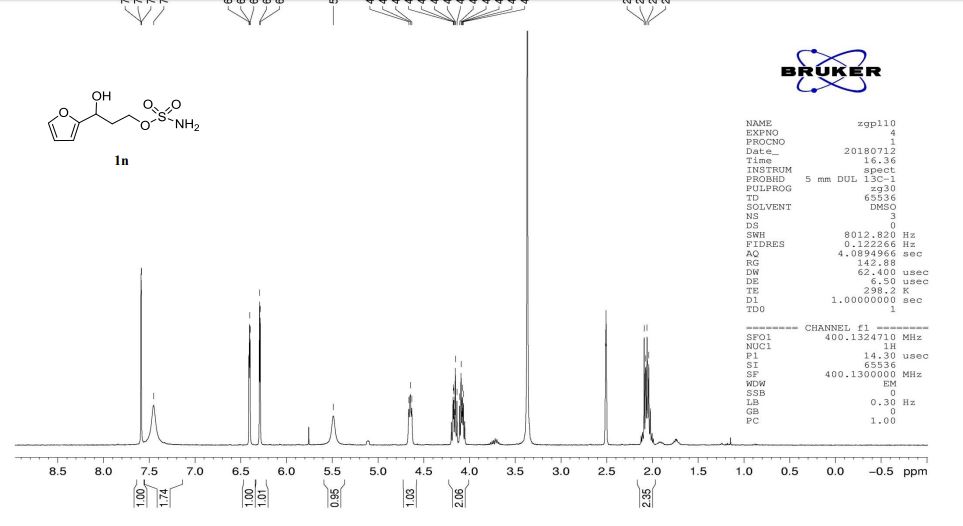
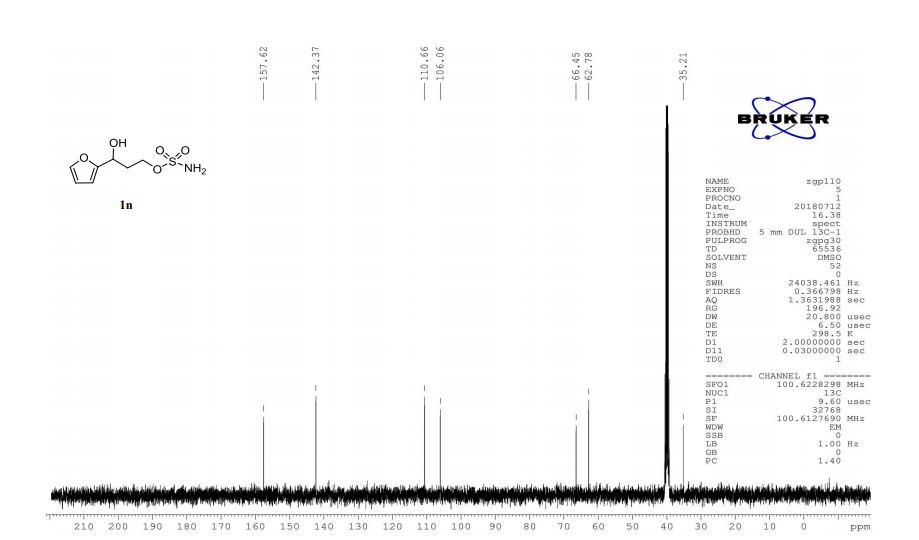
1n: colorless oil, 0.33 g, 73% yield for 2 steps.
1H-NMR (400 MHz, DMSO) δ: 7.59–7.58 (m, 1H), 7.45 (s, 2H), 6.40 (dd, J = 3.2, 1.8 Hz, 1H), 6.29 (d, J = 3.2 Hz, 1H), 5.49 (s, 1H), 4.74–4.60 (m, 1H), 4.18–4.07 (m, 2H), 2.09–2.04 (m, 2H).
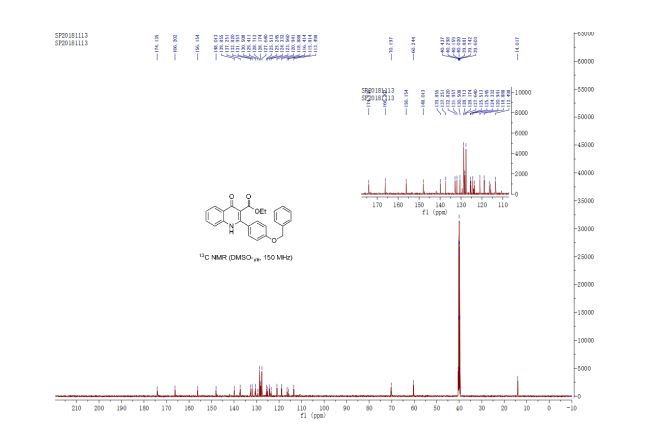
13C-NMR (100 MHz, DMSO) δ: 157.6, 142.4, 110.7, 106.1, 66.5, 62.8, 35.2. IR (KBr) 3282.9, 2928.7, 1627.4, 1562.5, 1353.8, 1174.6, 1074.0, 999.7, 918.4, 742.8 cm-1 ;
HRMS (CI+ ) (m/z) calcd. for C7H11NO5S [M]+ 221.0352; found 221.0354.
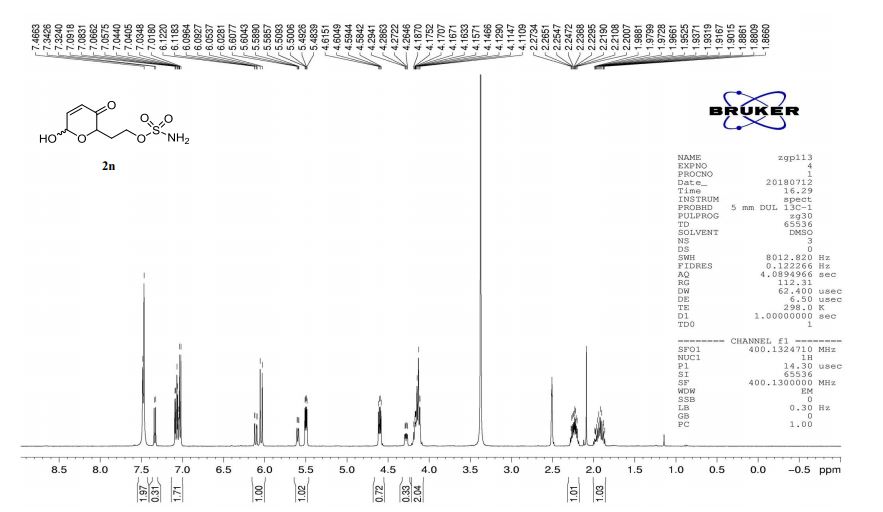
2n (EtOAc/hexane = 3:1):colorless oil (dr 7:3), 46 mg, 97%.
1H-NMR (400 MHz, DMSO) δ: 7.48–7.47 (m, 2H), 7.34–7.02 (m, 2H), 6.12–6.03 (m, 1H), 5.61–5.48 (m, 1H), 4.60 (dd, J = 8.3, 4.1 Hz, 0.7H), 4.28 (ddd, J = 8.8, 4.0, 1.3 Hz, 0.3H), 4.20–4.11 (m, 2H), 2.27–2.20 (m, 1H), 1.97–1.86 (m, 1H).
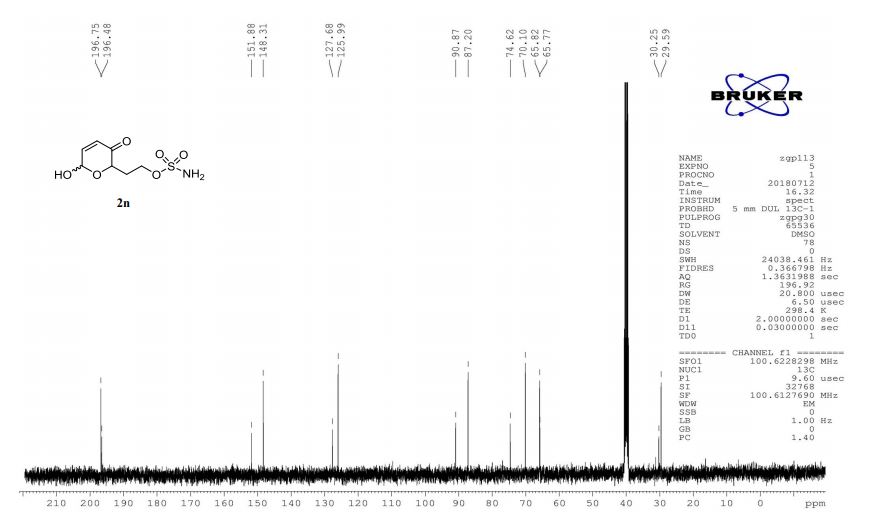
13C-NMR (100 MHz, DMSO) δ: 196.7, 196.5, 151.9, 148.3, 127.7, 126.0, 90.9, 87.2, 74.6, 70.1, 65.8, 65.8, 30.3, 29.6. IR (KBr) 3370.4, 2987.0, 1689.5, 1364.3, 1268.0, 1178.4, 1023.3, 928.3, 755.1 cm-1 ;
HRMS (CI+ ) (m/z) calcd. for C7H11NO6S [M]+ 237.0302; found 237.0315.

 *a
*a 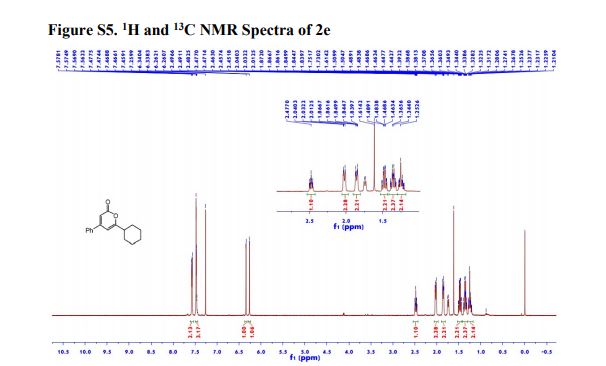
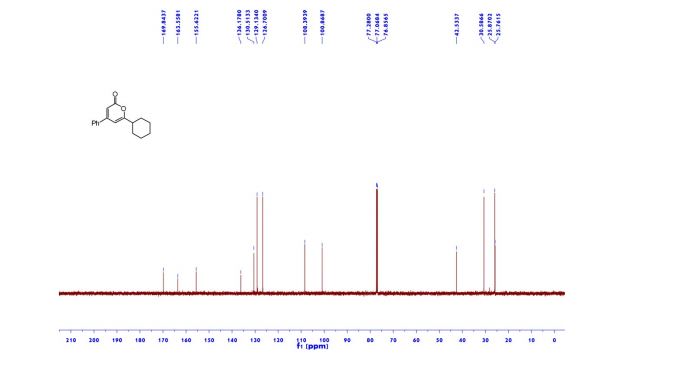


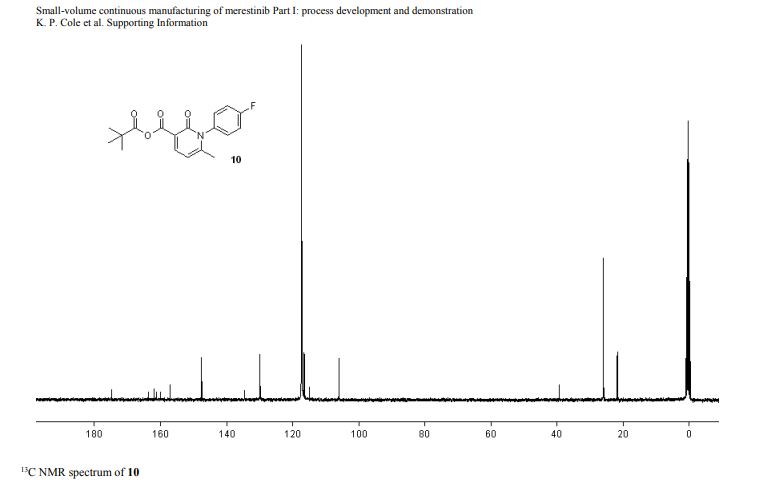
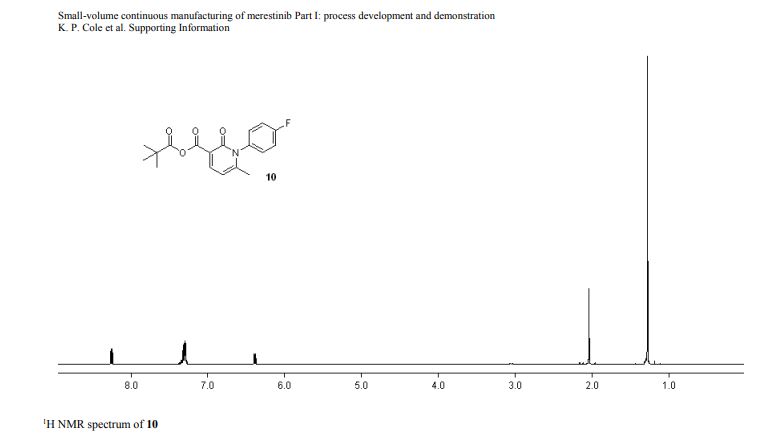



 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..



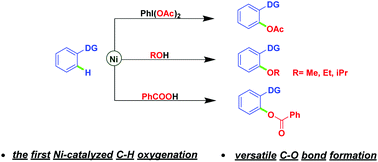

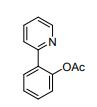
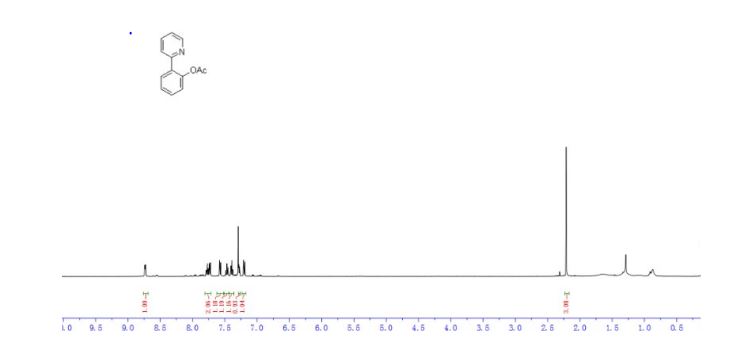
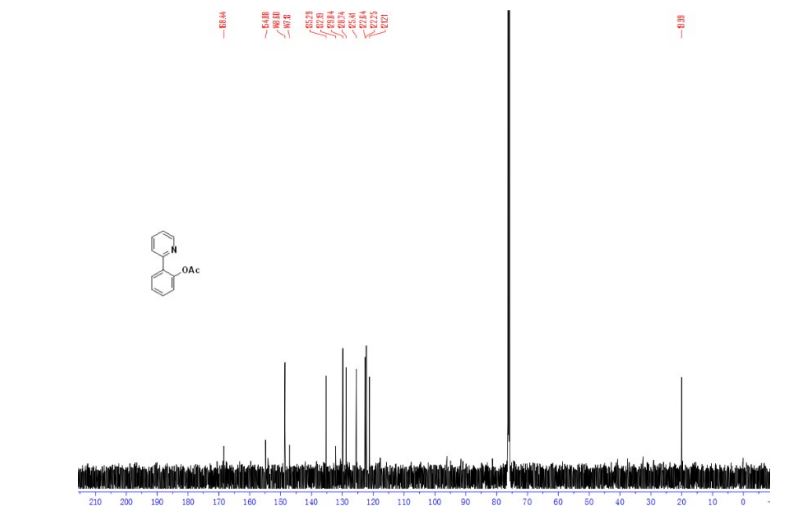
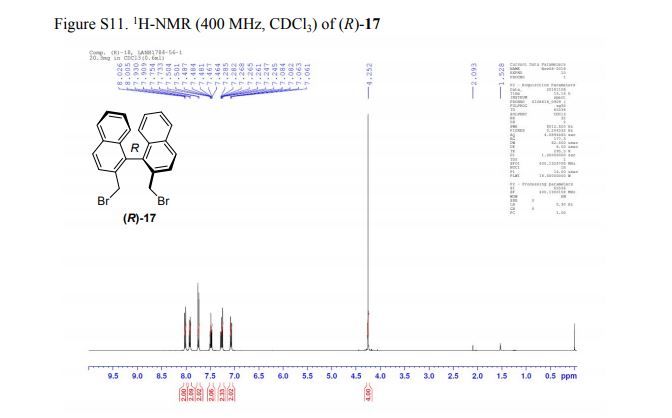
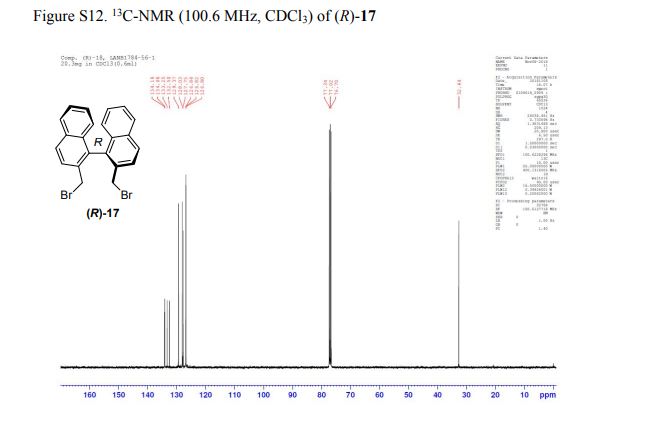



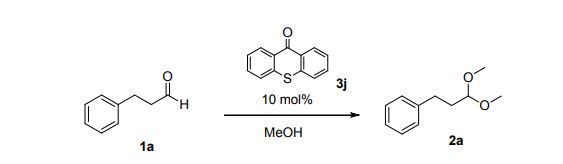

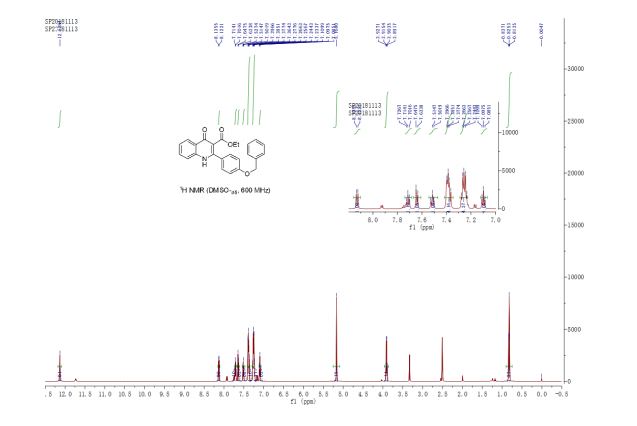
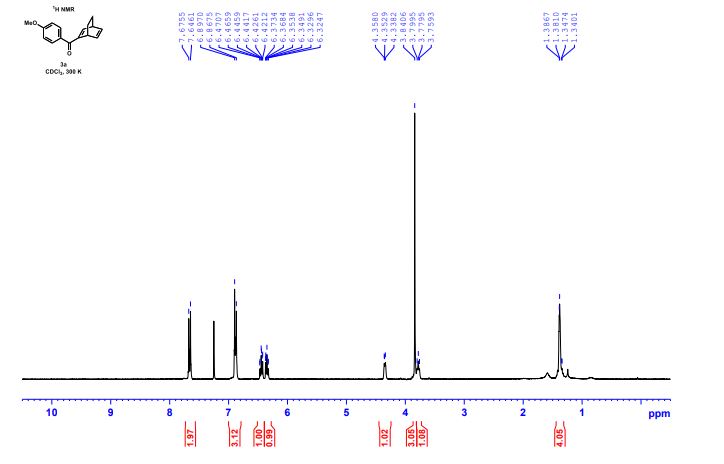
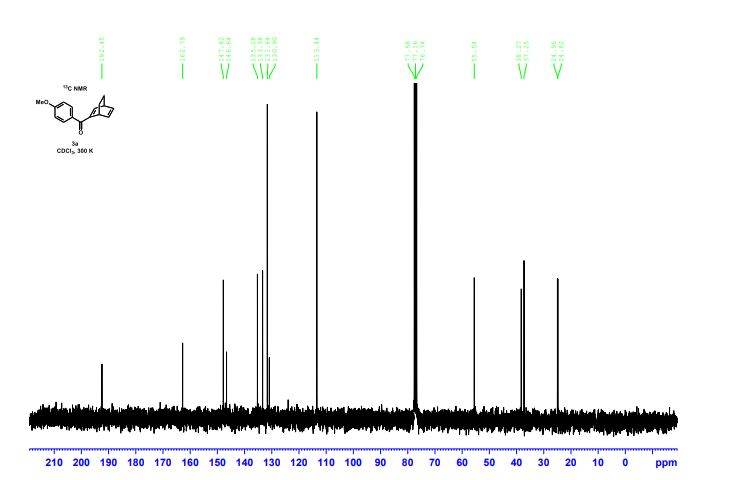
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....