
IRBESARTAN, SR 47436, BMS-186295
Avapro® (Bristol-Myers Squibb) and Karvea®
(Sanofi-Winthrop)
(Sanofi-Winthrop)
2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one
138402-11-6 CAS NO
U.S. Patents 5,270,317 and 5,352,788, 6,162,922
The compound prepared according to US 5270317 is polymorph A
- Irbesartan is known by following chemical names:
- (a) 2-Butyl-3-[[2'-(1H-tetrazol-5-yl)[1,1'-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (b) 2-Butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (c) 2-n-butyl-4-spirocyclopentane-1-[(2'-(tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one.
- The structural formula of Irbesartan is represented below.
Irbesartan
- The synthesis of irbesartan is first disclosed in US5270317 (equivalentEP0454511 ) and subsequently, several other patents disclose the synthesis of irbesartan by different methods. Basically the synthesis of this molecule involves two common intermediates namely spiroimidazole and substituted 4'-bromomethylbiphenyl.
- US 5270317 describes preparation of irbesartan wherein 1-[(2'-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin -5-one which is reacted with tributyltin azide in xylene at reflux temperature for 66 hours to give a product which is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol/THF mixture using 4N hydrochloric acid to get irbesartan.
- US5629331 describes a process for the preparation of irbesartan from 1-[(2'-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
Irbesartan (INN) /ɜrbəˈsɑrtən/ is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under thetrade names Aprovel, Karvea, and Avapro.
It is marketed in Brazil by Sanofi-Aventis under the trade name Aprovel .

As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
Irbesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Irbesartan/hydrochlorothiazide combination preparations are marketed under similar trade names to irbesartan preparations, including Irda, CoIrda, CoAprovel, Karvezide,Avalide and Avapro HCT.
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to placebo.[3]

BMS annual sales approx $1.3bn. Sanofi-aventis annual sales approx $2.1bn. In the United States, a generic version is available. Patent expired March 2012.
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. (2001). "Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes". N Engl J Med 345 (12): 851–60. doi:10.1056/NEJMoa011303.PMID 11565517.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A (December 2008). "Irbesartan in patients with heart failure and preserved ejection fraction". N. Engl. J. Med. 359 (23): 2456–67.doi:10.1056/NEJMoa0805450. PMID 19001508.
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5.... K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6... C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
Irbesartan of formula (I).



5,270,317, in the presence of an inert solvent such as DMF, DMSO or THF, with a basic reagent, for example KOH, a metal alcoholate, a metal hydride, calcium carbonate or triethylamine. The products of the reaction were purified by chromatography.
U.S. Pat. Nos. 5,352,788, and 5,559,233, and WO 91/14679 also describe identical alkylation of the nitrogen atom of the heterocyclic compound with the halo-biphenyl compound using the same inert solvent and the same basic reagents.
- US5629331 describes a process for the preparation of irbesartan from 1-[(2'-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
- WO 2005/051943 A1 describes a process for the preparing irbesartan wherein 1-[(2'-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with tributyltin chloride, sodium azide and TBAB in toluene at reflux temperature for 20 hours. Product is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol and formic acid to get irbesartan.
- WO 2006/023889 describes a method for preparing irbesartan, wherein 1-(2'-cyanobiphenyl-4-yl)methyl)-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with sodium azide and triethylamine hydrochloride in N-methyl-2-pyrrolidone to give irbesartan.
- WO 2005/113518 describes a process for preparing irbesartan wherein cyano irbesartan in xylene, is reacted with tributyltin chloride and sodium azide at reflux temperature till reaction is completed followed by aqueous work-up and recrystallization to give irbesartaN
- The process involving use of zinc salt for the transformation of nitrile to tetrazole is a safe and efficient process as reported in JOC (2001) 66, 7945-50. The use of zinc salt for transforming nitrile to tetrazole has also been published in WO9637481 and US5502191
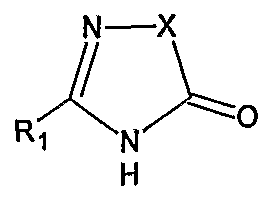

All of the above identified patents describe alkylation in solvents, such as N5N- dimethylformamide or DMSO, etc. in the presence of a basic reagent, for example, a metal hydride or a metal alcoholate etc. The strong bases, such as metal hydride or a metal alcoholate require anhydrous reaction conditions. Since N,N-dimethylformamide is used as a solvent, its removal requires high temperature concentration by distillation, which can result in degradation of the final product. The product intermediate is also purified by chromatography which is commercially not feasible and cumbersome on large scale. Another process given in Canadian Patent No. 2050769 provides synthetic scheme as herein given below.

US Patent No. 2004242894 also discloses the process of preparation of lrbesartan from 4- bromomethyl biphenyl 2'-(lH-tetrazol (2-triphenylmethyl) 5-yl) and Ethyl ester of 1- Valeramido cyclopentanecarboxylic acid in toluene in presence of base and PTC, and then hydrolyzing the protecting group. However this requires chromatographic purification.
This patent also discloses the process of preparation of tetrazolyl protected lrbesartan using 2,6 lutidine and oxalylchloride in toluene. However in this process the yield is as low as 30%.
US Patent No. 2004192713 discloses the process of preparation of lrbesartan by condensing the two intermediates via Suzuki coupling reaction. The reaction scheme is as given herein below.

WO2005113518 discloses the process of preparation of Irbesartan by condensing n- pentanoyl cycloleucine (V) with 2-(4-aminomethyl phenyl) benzonitrile (VI) using dicyclocarbodiimide (DCC) and 1 -hydroxy benzotriazole as catalyst to give an open chain intermediate of formula (VIII) which is then cyclized in the presence of an acid, preferably trifluoro acetic acid to give cyano derivative of formula (VII) and which in turn is converted to Irbesartan by treating it with tributyl tin chloride and sodium azide.

- Irbesartan is described in Bernhart et al., U.S. Patent No. 5,270,317
- Irbesartan, is a potent, long-acting angiotensin II receptor antagonist which is particularly useful in the treatment of cardiovascular ailments such as hypertension and heart failure. Its chemical name is2-n-butyl-4-spirocyclopentane-1-[(2'-(tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one.
According to EP 454511 a solid composition in form of tablets is prepared by mixing the active ingredient with a vehicle such as gelatine, starch, lactose, magnesium stearate, talc, gum Arabic or the like and can be optionally coated. The compositions containing from 20% to 70% by weight of irbesartan are known from EP 747050.
WO 04/007482 teaches the acidification to pH 2 - 3,5 of trityl irbesartan, which is sufficient to remove the protecting group, but not to convert into an acid addition salt; WO 04/065383 is likewise silent on hydrohalide acid addition salts. WO
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
...................................................
.....................

WO2006023889A2
Example 1Preparation of Compounds of formula IVa and IVb:

- A jacketed 1,000 mL 3-neck flask was charged with 4'-methylbiphenyl-2-carbonitrile (Compound 1, 100.0 g) and CH2CI2 (500 mL) under nitrogen. To a 500 mL Erlenmeyer flask with magnetic stirrer, sodium bromate (NaBrO3; 31.2 g) was dissolved in water (170 mL). The NaBrO3 solution was transferred to the 1,000 mL flask and the reaction mixture was cooled to about 5 °C or less. Aqueous HBr solution (48 %, 105.0 g) was added to the 1,000 mL flask and the resulting reaction mixture was recycled though a UV lamp reactor. The reaction mixture was kept at 0-20 °C and the recycling was continued until the reaction was deemed complete by HPLC. Optionally, additional sodium bromate and hydrogen bromide may be added. The relative amounts of Compound 2 and Compound 3 were about 80-90% and about 10-20% respectively. Aqueous sodium metabisulfite solution (2.0 g of in 10 mL water) was added to the reaction mixture. Allow the phases to settle and the methylene chloride phase was washed with water and used in the next step without further purification.

- A 1L 3-neck flask was charged with Compound V (134.0 g), MTBAC (5.0 g) and CH2Cl2 (170 mL) and cool to -5 to 5 °C. An aqueous solution of KOH (182.6 g in 212 mL water) was added slowly to the 1L flask and the reaction temperature was kept at ≤ 5 °C. The methylene chloride solution of Compound IVa and Compound IVb from Example 1 was added to the reaction mixture slowly, while maintaining the temperature at 0-10 °C. Diethyl phosphite (39.66g) was added drop wise at 0-10 °C. Check the reaction mixture for completion of the reduction reaction, and additional diethyl phosphite may be added.
- The reaction mixture was allowed to warm to ambient (20-30 °C) and agitated until the reaction was deemed complete by HPLC. Water (150 mL) was added and the phases were separated. The organic layer was extracted with water (230 mL) and polish filtered.
- The methylene chloride (which contained the crude Compound II) was distilled off and exchanged with about 400 mL of methyl tert-butyl ether (MTBE) (optionally, the MTBE recycled from washing below can be used here). Upon cooling, crystallization occurred (optionally seeds were added) and after further cooling to below 25°C, crystals of Compound II were isolated, washed with MTBE and dried in vacuum at a temperature of less than 60°C. HPLC retention time: 18.126 min. Typically, the yield was about 85 to about 88%. Alternatively, IPA could be used as the crystallization and washing solvent
- Optionally, the solvent (i.e., MTBE or IPA) used to wash the crystals of Compound II above can be recycled and used to crystallize the crude Compound II in the next batch. Since the washed solvent contains Compound II as well as impurities, it was surprisingly found that the washed solvent can be recovered and used again in crystallizing the crude compound of formula II in the next batch without sacrificing its purity while increasing its yield.

- A reactor was charged with Compound II (1 kg), triethylamine chlorhydrate (0.713 kg), sodium azide (0.337 kg) and N-methyl pyrrolidinone (2.07 kg), and the reaction mixture was heated to about 122°C under stirring. After completion of the reaction as determined by HPLC, the reaction mixture was cooled to about 45°C, and an aqueous solution of sodium hydroxide (35%, 5.99 kg) and water (3.0 kg) were added, the resulting mixture was stirred at a temperature between about 20 and about 40°C for about 0.5 hours. The aqueous phase was discarded and the organic phase was treated with toluene (1.73 kg) and water (5.0 kg), and stirred for about 0.5 hours at about 20 - about 30°C. The toluene phase was discarded and the aqueous phase was washed with ethyl acetate (1.8 kg) and treated with aqueous HCl until pH was adjusted to about 4.8 - about 5.2. Precipitation occurred and the resulting suspension was stirred for about 1 hour at about 20 - about 25°C. The precipitation was collected and washed with water three times (1.0 kg x 3). The crude wet product was recrystallized using a mixture of iso-propanol (0.393 kg) and water (4.5 kg). HPLC retention time: 11.725 min. The yield for Compound I was about 87%.
SPECTRAL DATA
The ESI mass spectrum of irbesartan showed a protonated molecular ion peak at m/z 429.3 confirming the molecular weight 428. The fragmentation pattern of parent ion 429.3 showed the fragment ions at m/z 385.9, 235.1, 207, 195.4, 192.1, 180.2 and 84
The FT-IR spectrum exhibited a characteristic stretching absorption band at 1732 cm-1 for the carbonyl group of amide functionality. The presence of this band at higher frequency was due to the ring stretching due to five member ring system. Another band at 1614cm-1 was due to C=N stretching vibrations
1H and 13C- NMR were recorded using DMSO-d6 as a solvent. In 1H-NMR the signal due to tetrazole NH proton was not detected may probably due to the tautomerism.
DP 1 IS IMPURITY
.................................................
NMR
WO2007049293A1
1H-NMR (DMSO d6): δppm 0.78 (t, 3H); 1.17-1.30 (sex, 2H); 1.40-1.50 (quent, 2H); 1.64-1.66 (m, 2H); 1.80-1.82 (m, 6H); 2.22-2.29 (t, 2H); 4.67 (s, 2H); 7.07 (s, 4H); 7.50- 7.68 (m, 4H) M+: 429.6
,.......................
m.p:181-182oC,
IR (KBr, cm-1) 1732 (C=O), 1616 (C=N); 1H NMR (DMSO-d6): δ 7.95–7.32 (m, 8 H), 4.80 –4.60 (s, 2 H), 3.60– 3.00 (br s, 1 H), 2.40– 2.20 (t, 2 H , J = 6.04 Hz), 2.00– 1.60 (m, 8 H),1.60–1.45 (quint, 2 H), 1.40– 1.20 (sext, 2 H), 0.91–0.70 (t, 3H, J = 7.41 Hz);
13C-NMR (DMSOd6): δ 186.5, 162.0,155.9, 141.9, 139.2, 137.2. 131.9, 131.4, 130.1, 128.7, 127.1, 124.3, 76.7, 43.1,
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
MS: m/z= 429 [M+1];
Anal. Calcd for C25H28N6O : C, 70.07; H,
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
http://www.acgpubs.org/OC/2011/Volume%204/Issue%201/13-OC-1106-199.pdf
............................
1H NMR in DMSO-D6 : 7.68 (d. 2H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.08 (s, 4 H, Ar-H), 4.68(s, 2H, -CH2), 2.69(t,2H,-CH2),2.18(m,2H,-CH2),1.83(m,2H,-CH2),1.81 (t, 2H, -CH2), 1.65 (t, 2H, -CH2), 1.45 (m, 2 H, -CH2), 1.24(m , 2H, -CH2), 0.77 (t, 3H, -CH3),
IR (KBR): 3061 (Aromatic C-H stretching), 2960 (Aliphatic C-H stretching), 3443 (N-H stretching), 1733 (C=0 stretching), 1617(CN stretching), 1337.99(CN stretching), 1407(N=N stretching) cm"1.
...................................................................
HPLC condition:
Column: Alltima C18 (Alltech 88050) 15.0cm in length x 4.6mm in internal diameter and 5 micron particle size;
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
The chromatographic purity of
the compounds was analyzed using Agilent 1200 series HPLC instrument under the following conditions:
Column : Symmetry C18, 4.6 × 75 mm, 3.5 µm
Mobile phase : Eluent A: Deionized water, Eluent B: HPLC grade Methanol
Chromatographic Conditions
a. Column temperature : Ambient
b. Sample compartment : Ambient
c. Detector : 225 nm
d. Injection volume : 10 µL
e. Run time : 45 minutes
f. Flow rate :1.0 mL/min
g. Injector :Auto sampler with variable volume injector
h. Diluent : HPLC grade Acetonitrile













