4-Iodobiphenyl 10 (420 mg, 1.5 mmol), Cy2NMe (0.39 mL, 1.8 mmol), Pd(OAc)2 (3 mg, 1 mol%) and tBu3PH.BF4 (9 mg, 2 mol%) were dissolved in PhMe/MeOH (5 mL, 9:1). The reaction mixture was injected into a Uniqsis Flowsyn reactor via a 5 mL PEEK injection loop. The reaction plug was pumped at 1.0 mL min−1 (using PhMe/MeOH (9:1) as stock solvent) through a tube-in-tube gas reactor pressurized with ethylene (15 bar) followed by a 20 mL PTFE reaction coil at 120 °C. The exiting reaction stream passed through an Omnifit column containing a mixture of QP-TU and QP-SA followed by a 200 psi BPR. The output was directed into a pre-weighed flask and flushed with argon.
The solvent was removed in vacuo to provide 268 mg (99%) of the title compound 11 as a white
crystalline solid.
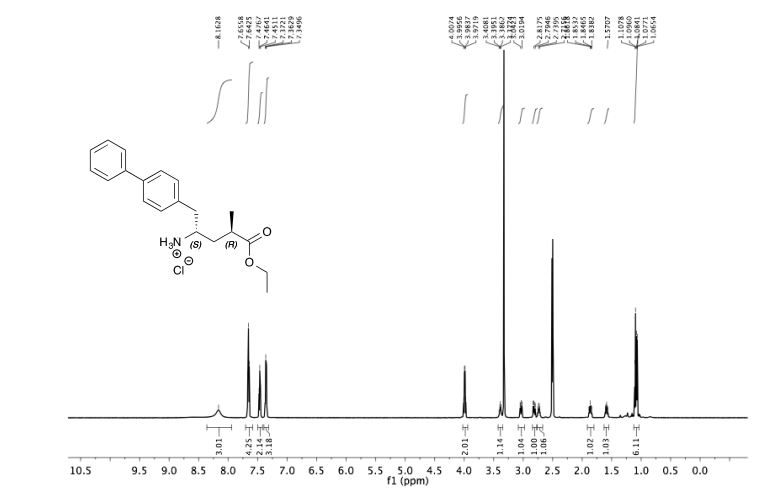
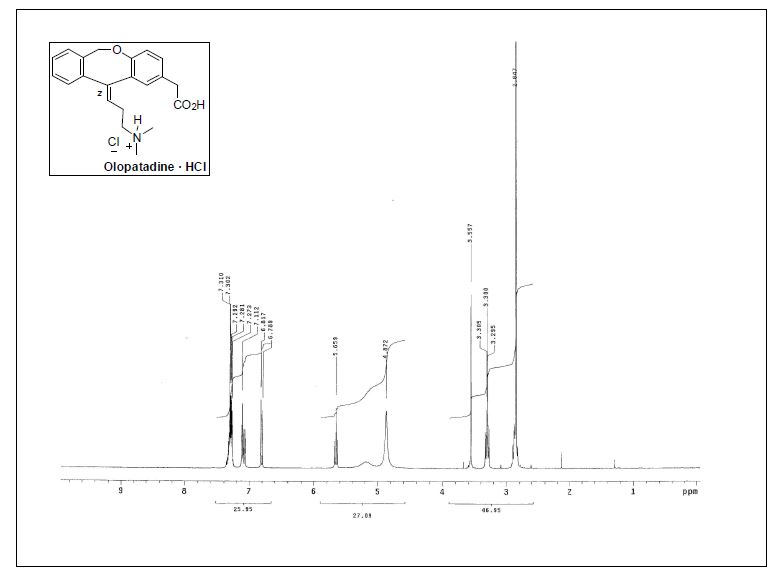
1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 7.2 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H),
7.48 (t, J = 7.4 Hz, 2H), 7.38 (t, J = 7.4 Hz, 1H), 6.78 (dd, J = 17.6, 11.0 Hz, 1H), 5.83 (d, J = 17.6 Hz, 1H),
5.31 (d, J = 11.0 Hz, 1H).
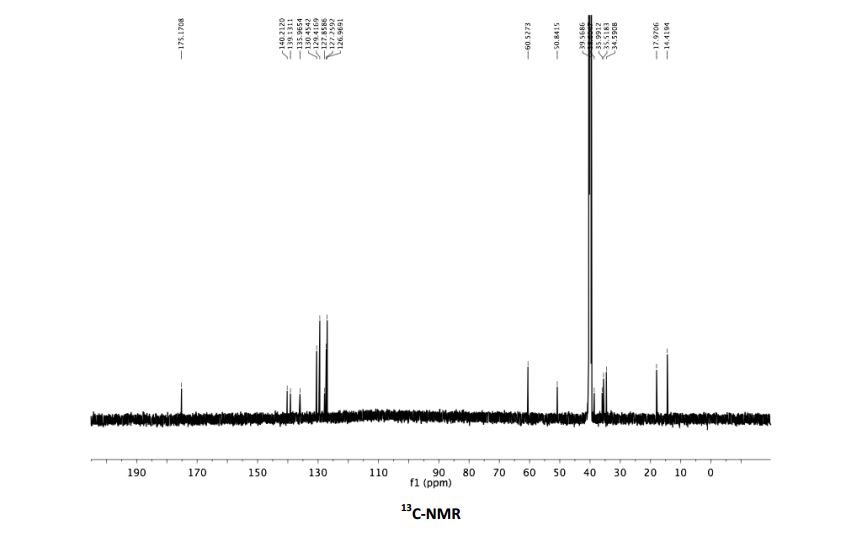
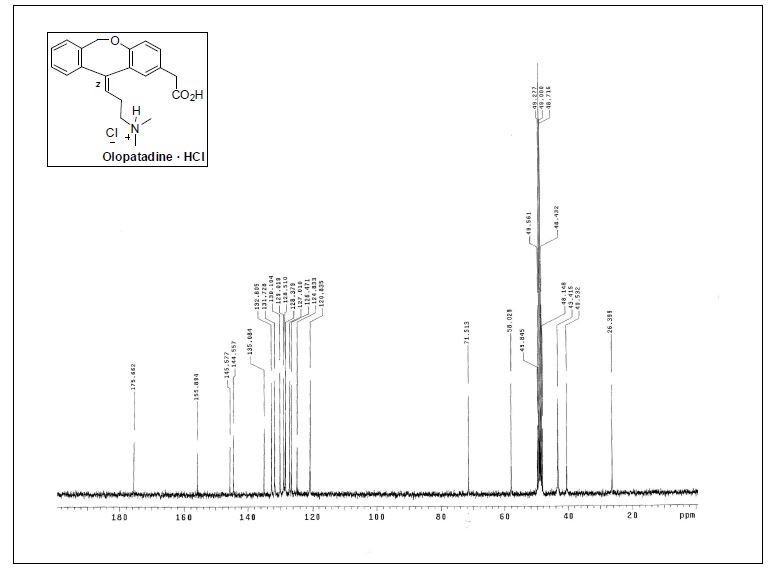
13C NMR (100 MHz, CDCl3) δ 140.7, 140.6, 136.6, 136.4, 128.7, 128.7, 127.3, 127.2, 127.2, 126.9,
126.9, 126.6, 126.6, 114.0.


///////////
Talgad Fort,
India, Maharashtra, Indapur, RAIGAD Kuda Mandad Caves, Ghosala fort
The fort was constructed during the fourth century by Raja Bhoj and was used as a watch-tower on Rajpuri river and creek.Those travelling from Roha to Murud via Tala can visit this fort which is just inside the main Tala village.








Talgad fort and Kuda- Mandad caves
After two successful, memorable monsoon treks at Kalsubai pinnacle and Sudhagad forts this season, Paulwaata group decided to plan another picnic cum trek to enjoy the rains on 21st of August 2011. The destination was Talgad Fort along with Kuda- Mandad Buddhist caves located near Roha. The trek was deliberately planned considering the easy nature of the same as there were going to be new comers with us which happened to trek for the very first time.
We left from Chembur by our private vehicle (Maruti- Omni) at around 6 in the morning. Weather seemed to be very pleasant with in-between showers. We deliberately took the Vashi- Chirner route for Pen as it passes through mountains and offers a great view. We moved steadily with photography sessions throughout our trip.

Our destination was village Tala situated at the base of the fort Talgad. This village can be approached either from Roha side or from taking a right turn from village Indapur (around 135 kms)on Mumbai- Goa highway NH-17. Other group members (Kedar and Co.) were supposed to come from Alibag by their Esteem car and thus we decided to gather at Roha first, have a heavy breakfast and then proceed towards Talgad. So we took the left turn before Nagothane village on Goa highway and moved towards Roha village about 20 kms from that spot. The road was in pathetic condition with big potholes for first few kms. Later on it was an enjoyable ride as the road passed through a small Ghat which looked magnificent in monsoons with heavy forest surroundings.
While moving towards Roha, few of kms prior to Roha station and railway crossings, we halted for a few minutes at village Medha which happens to be the base village of fort Avchitgad. Took some snaps of the fort and decided to visit it in near future. We reached Roha by around 9.30 and waited for the other group to come. Due to the poor road conditions, the other group came late and meanwhile we had our breakfast in a nearby Hotel Dwaraka. Samosa, Kachori, Vada paav and finally Glass full of divine Lassi! Could not have asked for anything better! Few members were having Fast since it was a Janmashtami day so we ordered special Upwasacha Chiwada and Lassi for them. Meanwhile Kedar and co. turned up so we moved towards Tala village (19 kms from Roha) after they finished with their breakfast by around 11.
As soon as we left Roha, rain started to pour like anything and heavy showers made the driving quite difficult. Roads are in decent conditions only in patches which slowed down our progress. Finally we reached the Tala village by 12, asked the villagers about the route to the fort. The village Tala is divided into two parts viz. Tala and Pusati wadi. The fort can be approached from both the sides but there is direct motorable road form Pusati wadi side which takes you almost at the foot the fort. Thanks to our vehicles, which managed those steep climbs, we could reach the spot in hardly 5 odd minutes. A small temple is there where you can observe few cannon lying. People in this part are very friendly and co-operative! From back of the village, a trail leads you into the jungle where there is thick bamboo growth. Just after 100 odd steps, a trail starts climbing towards right to the fort. It takes hardly ten minutes to reach the fort from this point. All the new comers thoroughly enjoyed this and the experienced ones were shocked to see how little they had to climb! On our way, great views of the surroundings were on offer and we made sure that we capture them in our cameras. There is statue of Hanuman carved in a big rock near the entrance of the door possible for guarding the fort and welcoming us! There is also a secret tunnel near the entrance which the villagers claim to be connected to the base village. Also visible are the remains of a large gateway and a water tank which has now been filled up.

Basic information about Talgad fort: A fortified monument on the southern bank of Malti creek, the Tala fort lies east west at a height of 1000 feet above the sea level in Tala town in Maharashtra. The fort is separated from the rest of the hill by a wide gap. This protected monument of 4th century was built during the reign of Bhoja Raja and was used as a watch on the beautiful Rajpuri stream. Tala fort is a very good example for the artistic talents of the then generation. The bottom of the hill in which the fort situates is surrounded by thick woody forest. Once a center of prime importance, this marvelous construction stands as the relic of its past glory.
The fort is rarely visited by trekkers and thus thankfully you don’t come across with plastic bags, bottles and scraps things like these on this fort! Hope it remains like this, untouched! After climbing the steps and entering the fort, we can see the main tower of the fort which is now in dilapidated condition and you can also see ruins of temple of Goddess Chandika near to that. We did lot of photography here. Great diversity of wild flowers can be seen on this fort and few members took snaps of them.

Then we started moving towards the other end of the fort where there is a bastion. En route we came across a series of water tanks covered with algal blooms suggesting the water was not potable. In all there are eleven water tanks. A small Shiva temple is next to the tanks.

After reaching the other end of the fort, the view was mesmerizing. Everyone wanted click snaps there and only after that we took some breather there and had mango drinks which we were carrying especially for the fasting people! We also had small introductory session here. Madhav had to respond to the call of the nature so he had vanished for few minutes!

It was already 2 pm by then so we decided to rush to the village as quickly as possible. Heavy non-stop pouring was making the descend very slippery. We got down in 20 odd minutes and moved towards Kuda- Mandad caves. To reach here, we took Tale- mandad road to the south of Tale village. The entire stretch from Talgad to the Kuda caves is a very interesting site and offers you great views of mountains and rivers. Mandad village is around 14 kms from Tala village. About few kms before Mandad, a kuccha road (Kuda Leni Phata) turns to the left and starts climbing the hill through the jungle. The motorable road is in poor condition so we moved as far as possible we can and then parked the vehicle and walked for next 10 odd mins.
Basic information about Kuda Caves:
Kuda caves, famous for Buddhist caves. These rock cut caves are classic example of Buddhist cave art and amazing for their architectural excellence. These caves have two levels and the individual blocks are smaller and are on the upper levels. The inscriptions on the caves are the preaching of Buddha and his disciples. The interiors of the caves are carved with stupas and a rock carved image of an elephant as the janitor is inscribed on the front gate of the caves. In Kuda caves, there are evidences to testify that monks used them as dwelling places. The inscriptions, letters & paintings in the Kuda hills shows that these caves are built in during first to sixth centuary B.C. 
There are a total of 13 caves in Kuda. Within the first cave, the walls of which are covered by ancient writing, is a stupa. This cave is much larger than the others. The sixth cave towards the left also has a stupa in it. This is the cave whose walls are adorned by elephants outside. There many bats inhabiting these beautiful caves so you prefer to enjoy them from outside only!

When you stand with your back towards the cave, Kuda village with the Murud creek in the backdrop is a clearly visible. This is great site especially in monsoons and the view is absolutely most picturesque.

We spent couple of hours here, took many photos and after changing in dry clothes (very much needed!) and having some CHYAU- MYAU tp food items, decided to move for our homes by 5.30 pm.
Considering the road conditions of towards Roha, we wisely decided to take Indapur road this time which is 15 kms from Tala village and said good byee to Kedar and co. who had to go to Alibag via Roha.
The road from Tala to Indapur is in excellent condition and we enjoyed this ride and wished had we come by same way in the morning we could have had more spare time and could have had visited Ghosalgad fort too! Nevertheless, whatever we had seen till now was quite refreshing and pleasant.

We set off from Indapur which is located on Mumbai- Goa highway by around 6.30 pm and moved towards Mumbai. We had our evening refreshments, tasty Misal paav, Potato chips at Hotel Kshudha Shanti at Wadkhal naaka at 8 pm. Finally reached Chembur by 10 pm. A very easy, non- strenuous enjoyable monsoon outing!
|




/////////////