![Propan-2-yl (2s)-2-[[[(2r,3r,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_sAO506pEhfr5XD7OmU3eafXuPVW7_xkfNECaBQ2Ig-6P-NjORR9fBn8Uj4d_g0PBn-pd3lcJNYGC-6ij8rr583-pJdqcN04zaODBN1S52NU9qadru8zcJDSIMxNA=s0-d)
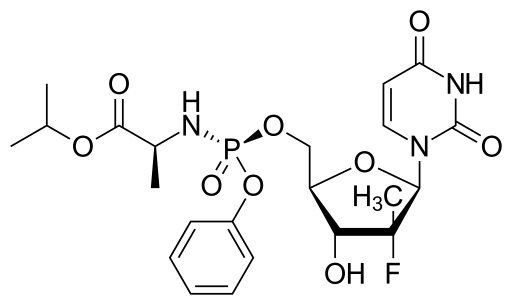
SOFOSBUVIR
SEE NMR PREDICTIONS BELOW
| Propan-2-yl (2s)-2-[[[(2r,3r,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate | |
| CAS No.: | 1190307-88-0 |
|---|---|
| Synonyms: |
|
| Formula: | C22H29FN3O9P |
| Exact Mass: | 529.16300 |
| Molecular Weight: | 529.45300 |
![propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate NMR spectra analysis, Chemical CAS NO. 1190307-88-0 NMR spectral analysis, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate H-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_t8qUqOISEVO1LMrLwnW4iJa6gpn8fAaUN5yzpfsgeBrB7wwCqk6cSN-dBwwl3ifIS4J5446LWZw-NrE-dQKqMuFUW9FmFrdAF6WpkE1jRmxQTYNEqeRwiDhbvs9fbQm-KCswcWQZxOsdRLfw=s0-d)
1H NMR PREDICTED
CAS NO. 1190307-88-0, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate H-NMR spectral analysis
![propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate NMR spectra analysis, Chemical CAS NO. 1190307-88-0 NMR spectral analysis, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate C-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_sJk9v_EDT8NTZ2MKkj61GCMRLKKxR9u18XR69KvdolrXBrmStvCEXJT7IVGEiExPqcGS5G2A2B89As0o2-UAjIxh6N_s0iGlXwJEnhiFLI0Bb3bvyQt426NeIpqA21YR-xcZqCCVMPdtxW9IM=s0-d)
13C NMR PREDICTED
CAS NO. 1190307-88-0, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate C-NMR spectral analysis
..............................................................................................
J. Med. Chem. 2010, 53, 7202–7218 DOI: 10.1021/jm100863x
http://www.chem.wisc.edu/deptfiles/chem345-gellman/Sp13/Antiviral_Drug/Sofosbuvir%20discovery%2010.pdf
51 (HPLC purity 99.74%, mp 93.9-104.7 C) in 15.2% yield from 6
.......................................................
NMR
http://file.selleckchem.com/downloads/nmr/S279401-Sofosbuvir-PSI-7977-HNMR-Selleck.pdf
......................................
http://www.google.co.in/patents/WO2013040492A2?cl=en
Example 10: Preparation of Compound 10 (from US2010/0298257)

phenydichlorophosphate (7.5 mL, 50 mmol) at room temperature. The mixture was cooled to - 10° C. and then was added a solution of N-Methylimidazole (30.5 mL, 384.3 mmol) in 30 mL of dichloromethane over a period of 30 min. After completion of the addition, the mixture was stirred between -10 and -15° C. for 1 h. To the above mixture was added 2'-deoxy-2'-fluoro-2'- C-methyluridine (10 g, 38.4 mmol) (see US2010/0298257 Example 1) in one lot and the mixture was stirred below -10° C. for 3 h and then slowly allowed to warm to 20° C. (6 h). The mixture was stirred at this temperature overnight (15 h) and then quenched with 10 mL of methanol. The solvent was evaporated and the residue was re-dissolved in EtOAc (200 mL). The EtOAc layer was washed with water (100 mL), 1 N HCI (3x75 mL), 2% aqueous NaHC03 solution (50 mL) and brine (50 mL). The organic layer was dried over Na2S04 filtered and concentrated. The residue was dried under high vacuum for 2 h to give white foam (22 g).
The above foam was dissolved in 33 mL of HCI and then was added 65 mL of isopropyl ether to give a saturated solution. The solution was filtered though a small pad of Celite and the filtrate was stirred with seeds of Compound 10 for 72 h at ambient temperature (about 22° C.-note that cooling the suspension to 0° C. led to oiling out the crude product). The white solid was filtered, washed with isopropyl ether (20 mL) and dried to give 4.58 g (-85:15 mixture of Compound 10: R isomer at P respectively as determined by 31 P NMR) of a white powder. The above solid was suspended in 23 mL of HCL and then refluxed for 3 h. The mixture was cooled to room temperature and stirred for 15 h. The white solid was filtered, washed with 4.5 mL of cold HCI and dried under high vacuum at 45° C. to give pure Compound 10, mp 93.9-104.7° C HPLC purity 99.74% (3.11 g, 15.2% from the uridine nucleoside).
Compound 10:
1H-NMR (CDCI3) δ 8.63 (br s, 1H, NH), 7.47 (d, 1 H, C6-H), 7.30 (m, 2H, o- aromatic), 7.26-7.18 (m, 3H, m,p-aromatic), 6.18 (br d, IH, Cl'-H), 5.70 (d, IH, C5-H), 5.02 (sept, CH-(CH3)2), 4.53 (m, 2H, C-5'-H2), 4.11 (d, IH, C3'-H), 3.97 (m, 3H, C3'-OH, C4'-H, ala-CH- CH3), 3.77 (br s, IH, ala-NH), 1.39 (d, 3H.C2'- CH3), 1.37 (d, 3H, ala- CH3) 1.24 (d, 6H, CH- (CH3)2).
................................
http://www.google.co.in/patents/US20100298257
Synthetic Aspects
In order to prepare the uridine nucleoside, one could take advantage of an advanced tribenzoylated cytidine intermediate in the synthesis of certain 3′,5′-diacylated analogs of 3 (see below) already produced efficiently on a pilot plant scale (see WO 2006/031725 or US 2006/0122146, both of which are incorporated by reference in their entirety). The following method was found to be scalable and cost-efficient.

Numerous literature procedures detail different routes and conditions to make phosphoramidates using several fold equivalents of reagents. See, for example, McGuigan et al. J. Med. Chem. 2005, 48, 3504-3515 and McGuigan et al. J. Med. Chem. 2006, 49, 7215. For process scale work, there is only one presently known example, which is disclosed in Lehsten et al., Org. Process Res. Dev. 2002, 6, 819-822 (“Lehsten”). In this reference, the authors introduce the concept of a “one-pot procedure” in which an amino acid hydrochloride salt and phenyl dichlorophosphate are reacted together with N-methylimidazole in dichloromethane. Later the nucleoside is added to form the desired 5′-O-phosphoramidate product, which in the present case would yield a compound represented by formula 4. Unfortunately, the Lehsten procedure suffered from drawbacks. For example, the Lehsten procedure utilized a far larger excess of reagents than was necessary which added to the cost and difficulty of chromatographic purification. Furthermore, Lehsten suggested that one could control the reaction selectivity on the 5′-hydroxyl over the 3′-hydroxyl compared to a literature reference through using lower temperatures and slow addition of the nucleoside.

Disclosed herein are reaction conditions which use lesser amounts of reagents and a method to selectively remove the impurity 3′-O-phosphoramidate diastereomers (5) with an easier chromatographic separation thereby affording the desired 5′-O-phosphoramidate diastereomers in much higher purity (4).
For the reagent stoichiometry, a study was made in which the stoichiometry of the reagents was systematically changed and the results were monitored by phosphorus NMR of the crude reaction as Lehsten had reported. In the more successful runs, the isolated yield and purity of the desired product were compared. It was observed that the primary 5′-hydroxyl reacts at a faster rate than the secondary 3′-hydroxyl. This creates a competing situation between the reaction progress of consuming all the starting nucleoside and converting 5′- and 3′-monosubstituted products (4 and 5) to the 5′,3′-bis substituted products (6). The 3′-monosubstituted product converts to the bis product at a faster rate than the 5′-monosubstituted product, so it is possible to reduce the 3′-diastereomer contamination level by pushing the reaction more to the bis-substituted products. However, with an effective way to remove the 3′-diastereomers, the reaction can be optimized to produce more of the desired 5′-diastereomer without having to sacrifice as much of the 5′-diastereomer being converted to the bis-substituted (6). It was also observed that the amino acid hydrochloride is very hygroscopic. As any water present would consume an equivalent amount of the phenyl dichlorophosphate reagent, care must be taken to keep the amino acid substantially anhydrous or it should be made substantially anhydrous prior to use. In short, Lehsten had reported that the optimum ratio of amino acid to phenyl dichlorophosphate to nucleoside was 3.5:2.5:1 respectively. It was found that the optimum ratio of amino acid to phenyl dichlorophosphate to nucleoside of about 1.6 to about 1.3 to about 1 is optimal under conditions in which the 3′-diastereomer can be efficiently removed and when the amino acid hydrochloride is substantially anhydrous. By using a smaller amount of the reagents, a cost savings is realized coupled with a simplification of the chromatographic separation of the desired product from reagent by-products and from the reduced level of bis diastereomers.
In one alternative procedure, a 3′-hydroxy-blocked derivative of 3 was prepared using a t-butyldimethylsilyl blocking group in two steps. This was then converted to its 5′-phosphoramidate derivative. The desire being that the silyl group could then be removed and there would be no 3′ isomers (5) or 3′,5′-bis phosphoramidates (6). A similar approach was demonstrated by Borch and Fries (U.S. Pat. No. 5,233,031) in a low overall yield on an alkyl phosphoramidate.
Another alternative approach was to use the direct synthesis and then use chemistry to help differentiate the 3′-diastereomer impurities 5 from the desired 5′-diastereomers 4 to help the separation. A group was desired that would selectively react with the free primary hydroxyl of the 3′-O-phosphoramidate impurity 5 over the free secondary hydroxyl of the desired 5′-O-phosphoramidate 4. It was also desired that the blocking group significantly change the polarity of the resulting 5′-O-blocked 3′-O-phoshoramidate product from the desired 5′-O-phosphoramidate 4. There would be no extra step needed to remove the blocking group as the desired 5′-diastereomers 4 would not be changed. The chemically altered 3′-diastereomers would then allow easier chromatographic separation or separation by special scavenging supports or by extractions.
Specifically, the blocking group tert-butyldimethylsilyl (tBDMS) met these criteria and was the first one to be demonstrated and subsequently used on a multi-kilogram scale. Under certain conditions such as in pyridine as solvent and base, the tBDMS group reacts with high selectively at the primary hydroxyl position over the 3′ secondary hydroxyl position. The phosphoramidate reaction uses N-methylimidazole (NMI) as a base. In the presence of NMI, the silylation is less selective. Preferably, the amount of NMI should be reduced. This can be accomplished easily after the phosphoramidate reaction by washing the reaction solution with 1 N hydrochloric acid. The NMI and the remaining starting nucleoside are removed, leaving a crude mixture of mono and bis substituted products and reagent by-products. This is then dissolved in pyridine and treated with tert-butyldimethylsilyl chloride. The 3′-monosubstituted product 5 is converted in a few hours or less to the 5′-O-tBDMS-3′-O-phosphoramidate 7. The reaction progress can be monitored by HPLC. The polarity of this silylated product 7 is less than the bis-phosphoramidate 6 and is readily removed by chromatography. Using this method, it was possible to reduce the level of 3′-monophosphoramidate 5 to less than 0.1% of the 5′-product 4 compared to 1-3% without the silyl treatment. Similarly, treatment with dimethoxytriphenylmethyl chloride (DMT-Cl) under the same conditions worked just as well. It was also easier to identify the DMT reaction product by TLC as DMT containing molecules stain bright orange on heating or exposure to acid. One can also envision many other blocking groups, as noted above.
Both the reaction conditions and the scavenging of the 3′-impurity are general methods and could be applied to most nucleoside phosphoramidates with a free 3′ hydroxyl. The phosphoramidate moiety could be any combination of amino acid ester and aromatic alcohol. The nucleoside moiety could be any nucleoside in which a 5′ phosphoramidate would lead to a 5′-monophosphate and could be further metabolized to the 5′-triphosphate form.
The following scheme is the main reaction scheme illustrated for making isopropyl L-alanate phenyl phosphoramidate of 2′-deoxy-2′-fluoro-2′-C-methyluridine with the major product as the desired 5′-O-phosphoramidate (4, two diastereomers) and the minor product as the 3′-O-phosphoramidate (5, two diastereomers) and the 3′,5′ -bis-O-phosphoramidate (6, four diastereomers). The reagents are added in the stoichiometric ratios as described in the method of preparation section. The reaction is allowed to proceed until about 5% of the starting material remains as judged by UV visualization on thin layer chromatography (TLC). Also UPLC/MS showed approximately 10% of the 3′,5′ bis-phosphoramidate 6 had formed compared to the desired 5′-product. After quenching and an acidic aqueous workup, the crude residue from the organic layer was prepared for the silylation. Under the described reaction conditions, the silyl group preferentially reacted with the free 5′-hydroxyl of the 3′-O-phosphoramidate to form 7. The reaction was continued until the 3′-O-phosphoramidate was no longer detectable by UPLC/MS.

Example 4 Preparation and Crystallization of SP-4
Method 1: Direct precipitation from crude 4: To a stirred solution of L-alanine isopropyl ester hydrochloride (10.5 g, 61.5 mmol, azeotropically dried, two times, with 50 mL of toluene each time) in dichloromethane (100 mL) was added phenydichlorophosphate (7.5 mL, 50 mmol) at room temperature. The mixture was cooled to −10° C. and then was added a solution of NMI (30.5 mL, 384.3 mmol) in 30 mL of dichloromethane over a period of 30 min. After completion of the addition, the mixture was stirred between −10 and −15° C. for 1 h. To the above mixture was added 2′-deoxy-2′-fluoro-2′-C-methyluridine (3) (10 g, 38.4 mmol) in one lot and the mixture was stirred below −10° C. for 3 h and then slowly allowed to warm to 20° C. (6 h). The mixture was stirred at this temperature over night (15 h) and then quenched with 10 mL of methanol. The solvent was evaporated and the residue was re-dissolved in EtOAc (200 mL). The EtOAc layer was washed with water (100 mL), 1N HCl (3×75 mL), 2% aqueous NaHCO3 solution (50 mL) and brine (50 mL). The organic layer was dried over Na2SO4, filtered and concentrated. The residue was dried under high vacuum for 2 h to give white foam (22 g).
The above foam was dissolved in 33 mL of DCM and then was added 65 mL of IPE (isopropyl ether) to give a saturated solution. The solution was filtered though a small pad of Celite and the filtrate was stirred with SP-4 seeds for 72 h at ambient temperature (about 22° C.—note that cooling the suspension to 0° C. led to oiling out the crude product). The white solid was filtered, washed with IPE (20 mL) and dried to give 4.58 g (˜85:15 mixture of SP-4:RP-4 respectively as determined by 31P NMR) of a white powder. The above solid was suspended in 23 mL of DCM and then refluxed for 3 h. The mixture was cooled to room temperature and stirred for 15 h. The white solid was filtered, washed with 4.5 mL of cold DCM and dried under high vacuum at 45° C. to give pure SP-4, mp 93.9-104.7° C., HPLC purity 99.74% (3.11 g, 15.2% from the uridine nucleoside).
SP-4 1H-NMR (CDCl3) δ 8.63 (br s, 1H, NH), 7.47 (d, 1H, C6-H), 7.30 (m, 2H, o-aromatic), 7.26-7.18 (m, 3H, m,p-aromatic), 6.18 (br d, 1H, C1′-H), 5.70 (d, 1H, C5-H), 5.02 (sept, CH—(CH3)2), 4.53 (m, 2H, C-5′-H2), 4.11 (d, 1H, C3′-H), 3.97 (m, 3H, C3′-OH, C4′-H, ala-CH—CH3), 3.77 (br s, 1H, ala-NH), 1.39 (d, 3H,C2′-CH3), 1.37 (d, 3H, ala-CH3), 1.24 (d, 6H, CH—(CH3)2).
updated
J. Med. Chem. 2005, 48, 5504.
WO2008045419A1
CN201180017181

UPDATE DEC 2015...........
SOFOSBUVIR
NEW PATENT WO2015188782,
(WO2015188782) METHOD FOR PREPARING SOFOSBUVIR
CHIA TAI TIANQING PHARMACEUTICAL GROUP CO., LTD [CN/CN]; No. 8 Julong North Rd., Xinpu District Lianyungang, Jiangsu 222006 (CN)
Sofosbuvir synthesis routes currently used include the following two methods:
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015188782&redirectedID=true

Preparation Example 1 sofosbuvir implementation
Step (a):
At
0 ℃, dichloro-phenyl phosphate (6.0g, 28.4mmol) in dry dichloromethane
(30ml) and stirred added alanine isopropyl ester hydrochloride (4.8g,
28.4mmol), the mixture After stirring and cooling to -55 ℃, was slowly
added dropwise triethylamine (6.5g, 64mmol) and dichloromethane (30ml)
mixed solution, keeping the temperature during at -55 ℃, dropping was
completed, stirring was continued for 60 minutes, after liters to -5 ℃
stirred for 2 hours, TLC monitored the reaction was complete. To remove
triethylamine hydrochloride was filtered and the filtrate evaporated
under reduced pressure to give compound 3-1 as a colorless oil (Sp / Rp =
1/1).
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 as internal standard): δ8.25 & 7.94 (1: 1);
1 HNMR (CDCl 3
, 300 MHz): δ7.39-7.34 (m, 2H), 7.27-7.18 (m, 3H), 5.10-5.02 (m, 1H),
4.51 (br, 1H), 4.11 (m, 1H ), 1.49 (d, 3H), 1.29-1.24 (m, 6H);
13 C NMR (CDCl 3 , 300 MHz): δ172.1 (Rp), 196.3 (Sp), 129.8,129.6 (d), 125.9,120.5 (d), 69.7 (d), 50.7 (d), 21.6 (d), 20.4 (d).
Step (b):
At
5 ℃, the compound of formula 2 (5.20g, 20.0mmol) in dry THF (30ml) and
stirred at t-butyl chloride (1.0M THF solution, 42ml, 42.0mmol). The
reaction temperature was raised to 25 ℃, and the mixture was stirred for
30 minutes. After addition of lithium chloride (21.0mmol), was slowly
added dropwise the compound 3-1 (approximately 28.4mmol) and THF (30ml)
mixed solution, keeping the temperature during at 5 ℃. Bi drops, stirred
for 15 hours. With aqueous 1N HCl (25ml) The reaction solution was
quenched (HPLC assay Sp: Rp ratio of 4: 1). Toluene was added (100ml),
temperature was raised to room temperature. The organic layer was washed
with 1N HCl, water, 5% Na 2 CO 3 and washed with
brine, dried over anhydrous magnesium sulfate, filtered, and the solvent
was distilled off under reduced pressure to a solid, was added
methylene chloride (20ml), stirred for 5 minutes plus isopropyl ether,
stirring was continued for 2 hours, the precipitated solid was filtered
off. The solid was dissolved by heating in dichloromethane (60ml),
slowly cooled to room temperature and the precipitated crystalline
solid. Repeat if necessary obtain pure crystalline sofosbuvir (2.6g,
yield 25%, HPLC purity measured 98.8%).
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 as internal standard): δ3.54ppm;
13 C NMR (CDCl 3 , 300 Hz): δ173.1 (d), 162.7 (s), 150.2 (d), 139.3 (d), 129.6 (q);
MS (M + H): 530.1.
Preparation of compounds of formula 2 shown in Example 3-2
(1) a nucleophilic reagent as NaSCN, the phase transfer catalyst is TBAB
The
compound (product of Example 1, step (a)) is represented by the formula
3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol),
the NaSCN (35mmol) in water (2.0ml) was added dropwise It was added to
the reaction solution. Dropping was completed, stirring was continued
for 60 minutes, the solid was removed by filtration. The filtrate was
washed with water, add MgSO 4 dried for 24 hours. Filtered,
and the filtrate was evaporated under reduced pressure, to obtain a
compound of formula 3-2 as (where X = SCN).
1 HNMR (CDCl 3 , 500Hz): δ7.32-7.13 (m, 3H), 7.08-7.02 (m, 2H), 5.0-4.9 (m, 1H), 3.92 (m, 1H), 1.49 (m, 3H ), 1.23-1.17 (m, 6H);
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ-18.16 / -18.26.
(2) nucleophile NaSCN, phase transfer catalyst is 18-crown-6 ether
The
compound (product of Example 1, step (a)) is represented by the formula
3-1 is dissolved in ethyl acetate (20ml) was added 18-crown -6
(2.8mmol), the NaSCN (35mmol) was added to the above the reaction
mixture. Dropping was completed, stirring was continued for 60 minutes,
the solid was removed by filtration. The filtrate was washed with water,
add MgSO 4 dried for 24 hours. Filtered, and the filtrate
was evaporated under reduced pressure, to obtain a compound of formula
3-2 as (where X = SCN).
(3) nucleophile NaSCN, phase transfer catalyst is TBAB and 18-crown-6
The
compound (product of Example 1, step (a)) is represented by the formula
3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol) and
18-crown -6 (2.8mmol), the NaSCN (35mmol) in water (2.0ml) was added to
the reaction solution. Dropping was completed, stirring was continued
for 60 minutes, the solid was removed by filtration. The filtrate was
washed with water, add MgSO 4 dried for 24 hours. Filtered,
and the filtrate was evaporated under reduced pressure, to obtain a
compound of formula 3-2 as (where X = SCN).
(4) nucleophile as NaN 3 , phase transfer catalyst is TBAB
The
compound (product of Example 1, step (a)) is represented by the formula
3-1 is dissolved in dichloromethane (20ml) was added TBAB (2.8mmol),
the NaN 3 (35 mmol) in water (2.0ml) solution of was added
dropwise to the reaction solution. Dropping was completed, stirring was
continued for 60 minutes, the solid was removed by filtration. The
filtrate was washed with water, add MgSO 4 dried for 24
hours. Filtered, and the filtrate was evaporated under reduced pressure,
to obtain a compound of formula 3-2 as (where X = N 3 ).
1 HNMR (CDCl 3 , 500Hz): δ7.30-7.33 (m, 2H), 7.27-7.21 (m, 3H), 5.10-5.05 (m, 1H), 4.12-4.00 (m, 1H), 1.43 (d , 3H), 1.28-1.17 (m, 6H);
31 PNMR- (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ2.04 / 2.19.
(5) the nucleophilic reagent is KCN, the phase transfer catalyst is TBAB
The
compound was dissolved in methylene chloride as in formula 3-1 (20ml),
was added TBAB (2.8mmol), the KCN (35mmol) in water (2.0ml) was added
dropwise to the reaction solution. Dropping was completed, stirring was
continued for 60 minutes, the solid was removed by filtration. The
filtrate was washed with water, add MgSO 4 dried for 24
hours. Filtered, and the filtrate was evaporated under reduced pressure
to remove the solvent to give a compound as shown in Formula 3-2 (where X
= CN).
1 HNMR (CDCl 3 , 300 Hz): δ7.22-7.13 (m, 3H), 7.09-7.02 (m, 2H), 5.01-4.95 (m, 1H), 4.08-3.93 (m, 1H), 1.43-1.35 (m, 3H), 1.20-1.17 (m, 6H);
31 PNMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ-2.71 / -2.93.
Preparation Example 3 sofosbuvir implementation
(1) X is SCN
Under
5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF
(30ml) in. T-butyl chloride was added with stirring (1.0M THF solution,
42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the
mixture was stirred for 30 minutes. After addition of lithium chloride
(21.0mmol), was slowly added dropwise a compound of formula 3-2
(Preparation Example 2 28.4 mmol, obtained) and THF (30ml) mixed
solution, keeping the temperature during at 5 ℃. After dropping was
completed, the mixture was stirred for 15 hours. With aqueous 1N HCl
(25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 6:
1). After further addition of toluene (100ml), temperature was raised
to room temperature. The organic layer was washed with 1N HCl, water, 5%
Na 2 CO 3 and washed with brine, dried over
anhydrous magnesium sulfate, filtered, and the solvent was distilled off
under reduced pressure to a solid, was added methylene chloride (20ml),
stirred for 5 minutes plus isopropyl ether, stirring was continued for 2
hours, the precipitated solid was filtered off. The solid was dissolved
by heating in dichloromethane (60ml), slowly cooled to room temperature
and the precipitated crystalline solid. Repeat if necessary obtain pure
crystalline sofosbuvir (3.6g, yield 34%, HPLC purity measured 98.7%).
1 HNMR (CDCl 3
, 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H,
O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H),
5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
(2) X is N 3
Under
5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF
(30ml) in. T-butyl chloride was added with stirring (1.0M THF solution,
42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the
mixture was stirred for 30 minutes. Was added lithium chloride
(21.0mmol), was slowly added dropwise after the compound of formula 3-2
obtained in Preparation Example 2 (about 28.4 mmol) and THF (30ml) mixed
solution, keeping the temperature during at 5 ℃. Bi drops, stirred for
15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched (HPLC assay Sp: Rp ratio of 7: 1). After
further addition of toluene (100ml), temperature was raised to room
temperature. The organic layer was washed with 1N HCl, water, 5% Na 2 CO 3
and washed with brine, dried over anhydrous magnesium sulfate,
filtered, and the solvent was distilled off under reduced pressure to a
solid, was added methylene chloride (20ml), stirred for 5 minutes plus
isopropyl ether, stirring was continued for 2 hours, the precipitated
solid was filtered off. The solid was dissolved by heating in
dichloromethane (60ml), slowly cooled to room temperature and the
precipitated crystalline solid. Repeat if necessary obtain pure
crystalline sofosbuvir (4.2g, yield 40%, HPLC purity measured 98.8%).
1 HNMR (CDCl 3
, 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H,
O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H),
5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
(3) X is CN
Under
5 ℃, the compound (5.20g, 20.0mmol) as shown in Equation 2 in dry THF
(30ml) in. T-butyl chloride was added with stirring (1.0M THF solution,
42ml, 42.0mmol). The reaction temperature was raised to 25 ℃, and the
mixture was stirred for 30 minutes. After addition of lithium chloride
(21.0mmol), was slowly added dropwise a compound of formula 3-2 obtained
in Preparation Example 2 (about 28.4 mmol) and THF (30ml) mixed
solution, keeping the temperature during at 5 ℃. Bi drops, stirred for
15 hours. With aqueous 1N HCl (25ml) The reaction solution was quenched
(HPLC assay Sp: Rp ratio of 6: 1). After further addition of toluene
(100ml), temperature was raised to room temperature. The organic layer
was washed with 1N HCl, water, 5% Na 2 CO 3 and
washed with brine, dried over anhydrous magnesium sulfate, filtered, and
the solvent was distilled off under reduced pressure to a solid, was
added methylene chloride (20ml), stirred for 5 minutes plus isopropyl
ether, stirring was continued for 2 hours, the precipitated solid was
filtered off. The solid was dissolved by heating in dichloromethane
(60ml), slowly cooled to room temperature and the precipitated
crystalline solid. Repeat if necessary obtain pure crystalline
sofosbuvir (4.02g, yield 40%, HPLC purity measured 98.8%).
1 HNMR (CDCl 3
, 300 MHz): [delta] 8.63 (s, 1H, NH), 7.46 (d, 1H, C6-H), 7.36 (t, 2H,
O-aromatic), 7.18-7.24 (m, 3H, m, P-aromatic), 6.20-6.14 (d, 1H, Cl'-H),
5.70-5.68 (d, 1H, C5-H), 5.05-4.97 (m, 1H, CH- (CH 3 ) 2 ) , 4.57-4.41 (m, 2H, C5'-H2), 4.12-4.09 (d, 1H, C3'-H), 4.06-3.79 (m, 3H, C3'-OH, C4'-H, Ala-CH -CH 3 ), 3.79 (s, 1H, Ala-NH), 1.44 (d, 3H, C2'-H3), 1.36-1.34 (d, 3H, Ala-CH 3 ), 1.25-1.23 (t, 6H, CH- (CH 3 ) 2 );
P 31 NMR (CDCl 3 , 300 Hz, H 3 PO 4 internal standard): δ3.56.
//////
 COCK WILL TEACH YOU
COCK WILL TEACH YOU





No comments:
Post a Comment