

Axitinib (AG013736; trade name Inlyta) is a small molecule tyrosine kinase inhibitor developed by Pfizer. It has been shown to significantly inhibit growth of breast cancer in animal (xenograft) models[2] and has shown partial responses in clinical trials with renal cell carcinoma (RCC)[3] and several other tumour types.[4] It was approved by the U.S. Food and Drug Administration after showing a modest increase in progression-free survival,[5] though there have been reports of fatal adverse effects.[6]
Axitinib, a small-molecule indazole derivative chemically known as (E)-N-methyl-2-(3-(2-(pyridin-2-yl)-vinyl)-1H-indazol-6-ylthio)benzamide developed by Pfizer, was approved in January 2012 by the U.S. FDA with the trade name Inlyta. It selectively inhibits vascular endothelial growth factor receptors for the treatment of renal cell carcinoma
On January 27, 2012, axitinib was approved with the trade name INLYTA for treatment of patients in the United States with advanced renal cell carcinoma after failure of one prior systemic therapy.
It has received FDA (27 January 2012), EMA (13 September 2012), MHRA (3 September 2012) and TGA (26 July 2012) approval for use as a treatment for renal cell carcinoma.[11][12][13][14]
A study published in 2015[15] showed that axitinib effectively inhibits a mutated gene (BCR-ABL1[T315I]) that is common in chronic myeloid leukemias and adult acute lymphoblastic leukemias which have become resistant to other tyrosine kinase inhibitors likeimatinib. This is one of the first examples of a new indication for an existing drug being discovered by screening known drugs using a patient's own cells.

The discovery and development of an efficient synthesis route to axinitib is reported. The first-generation route researched by Pfizer implemented two Pd-catalyzed coupling reactions as key steps. In this work, the development of Heck-type and C–S coupling reactions catalyzed by CuI is briefly described, using an economial and practical protocol. Aspects of this route, such as selecting optimal ligands, solvent, and other conditions, are discussed in detail. The scale-up experiment was carried out to provide more than 300 g of active pharmaceutical ingredients of axitinib in Form XLI with 99.9% purity in 39% yield. In short, we provide a new choice of synthesis route to axitinib, through two copper-catalyzed coupling reactions with good yield.
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00123
(E)-N-Methyl-2-(3-(2-(pyridin-2-yl)vinyl)-1H-indazol-6-ylthiol)benzamide
(Axitinib) Form XLI (326.4 g in 96% yield with purity 99.91%). Residual
Cu content was determined to be 2.2 ppm by atomic absorption
spectroscopy: mp 227.7 °C; 1H NMR (300 MHz, DMSO-d6) δ 13.27 (s, 1H), 8.60 (d, J = 4.8 Hz, 1H), 8.29 (d, J = 5.4 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 16.4 Hz, 1H), 7.81 (t, J = 7.5 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.63–7.44 (m, 3H), 7.29 (p, J = 7.4, 6.6 Hz, 3H), 7.19 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 7.4 Hz, 1H), 2.78 (d, J = 4.6 Hz, 3H);
13C NMR (75 MHz, DMSO-d6) δ 167.89, 154.86, 149.54, 142.01, 141.86, 136.92, 136.88, 135.67, 132.52, 130.32, 129.99, 129.25, 127.80, 126.15, 125.59, 123.66, 122.68, 122.50, 121.79, 120.29, 114.76, 26.13.
.............................
Axitinib (Axitinib, AG-013736, CAS: 319460-85-0) is a Pfizer research and development by the United States of new, mainly targeting VEGFR kinase GABA, inhibiting angiogenesis anticancer small molecule drug, trade name Inlyta, for other systems therapy for advanced renal cell carcinoma (Renal Cell Carcinoma, RCC), 2008 has been approved in the domestic clinical, and Pfizer's cancer drug Sutent another similar imatinib (Sunitinib) , Axitinib also potent and selective multi-targeted tyrosine kinase inhibitor, can inhibit the vascular endothelial growth factor receptor (Vascular EndothelialGrowth Factor Rec India tor, VEGFR), including VEGFl receptor, VECF2 receptors and VECF3 receptor, can inhibit platelet-derived growth factor receptor (Platelet-derived growth factor receptor, PDGFR) and c_KIT. Axitinib is called sunitinib second generation, better than sunitinib adverse reactions.
Axitinib (II) chemical name 6- [2_ (methylcarbamoyl) phenylsulfanyl] -3-E- [2_ (Batch-2-yl) ethenyl] indazole structural formula as follows:

Assi synthesis method for Nepal mainly in the following three ways:
(I) Patent US20060094881 (Agouron Pharmaceuticals), EP2163544 (Pfizer) reported the first synthesis method Axitinib to 3,6-diiodo-indazole as a starting material, first-iodo-6-position is substituted mercapto group, protection of the NH group, then the Heck reaction occurs (pyridine-2-yl) vinyl 3-position, after deprotection Axitinib whole synthesis route is as follows:

This method although the synthesis route is shorter, but the catalyst and reagents used relatively expensive and require purified through the column, the total yield is low, is not conducive to industrial production.
[0004] (2) The second method of synthesis Axitinib e.g. W00102369 (Agouron Pharmaceuticals), US6531491 (Agouron Pharmaceuticals) reported in 6-nitro-indazole as a starting material, the 3-position first iodo, followed by the protecting group NH, Suzuki coupling reaction with boronic acid to give 3- styryl styryl-position, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, the 3-position of the styrene-based ozone of the obtained aldehyde, followed by Wittig reaction to give the 3-position (pyridin-2-yl) ethenyl, 6-position is substituted mercapto iodine, alkaline hydrolysis then amidated, and finally deprotection Axitinib, the entire reaction formula as follows:

The method of synthesis route is long, harsh reaction conditions, complex process, the total yield is low, does not apply to industrial production.
[0005] (3) The third method is W02006048745 (Pfizer) discloses to 6-nitro-indazole as a starting material, the 3-position iodo first, followed by the protecting group NH, 3- bits Heck coupling reaction, a nitro group reduced to an amino group, an amino diazotization reaction was iodo, iodo-6-position is substituted mercapto group, and finally deprotected to give Axitinib, the entire reaction is as follows:
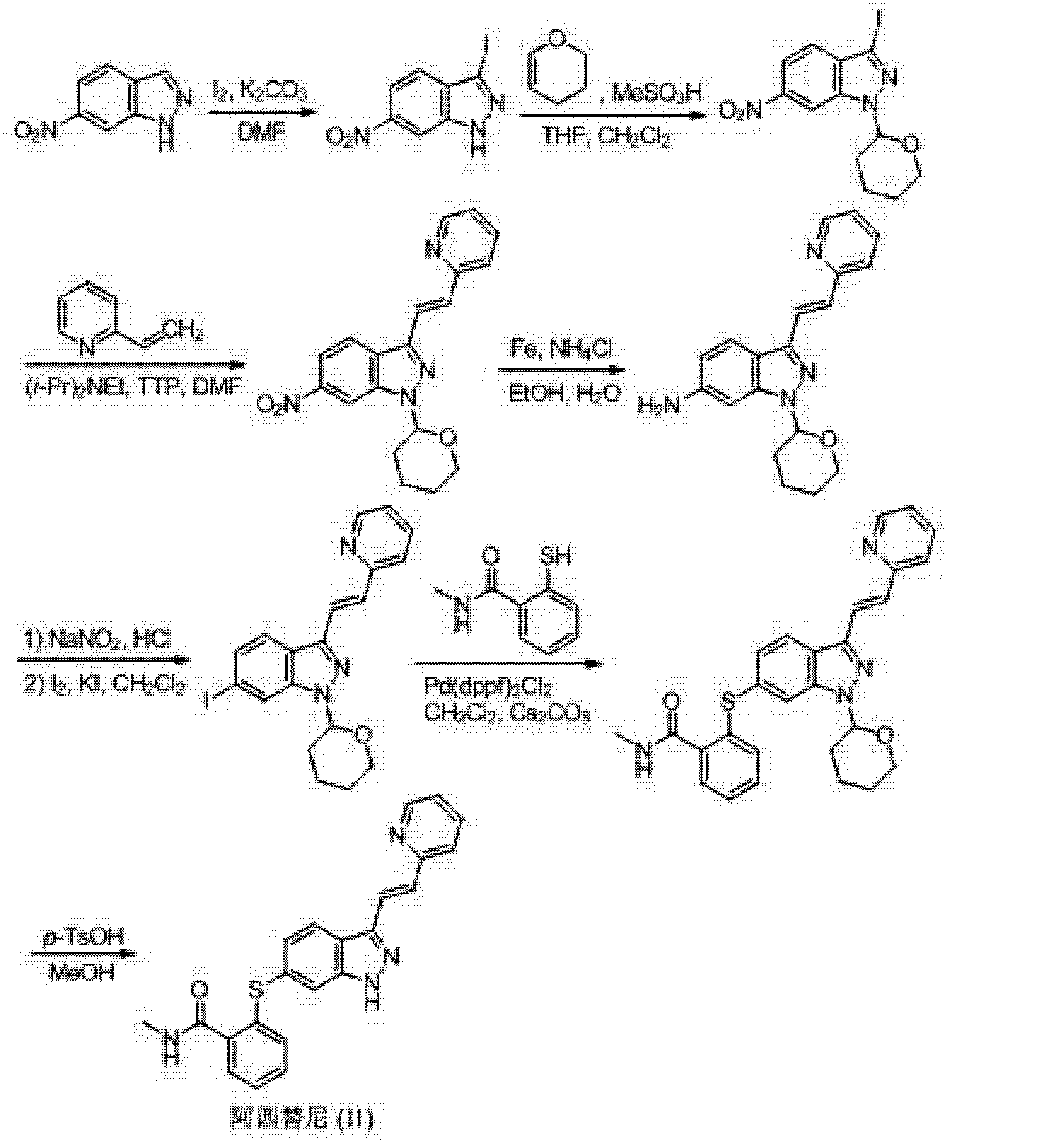
SYNTHESIS

aReagents and conditions: (a) I2, K2CO3, DMF; (b) CH2Cl2, CH3SO3H, dihydrofuran; (c) compound B, i-Pr2EtN, Pd(OAc)2, (o-Tol)3P, DMF; (d) iron, EtOH, NH4Cl; (e) AcOH, NaNO2, CH2Cl2, I2/KI; (f) compound C, Pd(dppf)Cl2, Cs2CO3, DMF; (h) 1, p-TsOH, MeOH; 2, NaHCO3; (i) AcOH, MeOH, Pd removal, recrystallization.
http://www.google.com/patents/WO2006048745A1?cl=en
Example 15: Final deprotectioπ step to produce 6-r2-(methylcarbamoyl)phenylsulfanyll-3-E-f2- (pyridine-2-yl)ethenyllindazole

Example 21 : Preparation of 6-F2-(methylcarbamovDphenylsulfanyll-3-Z-r2-(pyridine-2- vDethenyllindazole


Polymorph Form IV
5) HOAc/Xylenes
To a 12 L 3-neck flask, equipped with a mechanical stirrer, was added 160.20 g of 6-[2- (methylc'arbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole and 1.6 L of DMA and 1.6 L of THF. After stirring for 20 minutes, the mixture became homogeneous. To the clear solution was added 800.99 g of 10% cysteine-silica and the resulting mixture was allowed to stir at room temperature overnight.
The mixture was filtered through a medium sintered glass fritted funnel, and the cake was washed with a solution of 500 mL of DMA and 500 mL of THF. The cake was further washed with 2.0 L of THF and the filtrate was collected into a separate flask. The volatile parts in the latter filtrate were removed in vacuo and the residue was combined with the main filtrate. The combined filtrate was recharged back into the 12 L flask, followed by 800 g of 10% cysteine-silica. The flask was equipped with a mechanical stirrer and stirred over the weekend at room temperature. The mixture was then filtered through a medium sintered glass fritted funnel and the silica was washed with a mixture of solvents of 500 ml. of DMA and 500 ml_ of THF, followed by 3.0 L of THF. The volatile parts in the filtrate were removed in vacuo and the remaining solution was transferred to a 22 L 3-neck flask and treated with 12 L of water (added over a 20 minute period of time), a thick precipitate formed at this stage. After stirring overnight, the mixture was filtered and the cake was washed with 2.0 L of water and sucked dry.
The cake was charged to a 5 L 3-neck flask, followed by 1.6 L of THF and 160 mL of DMF. The flask was equipped with a mechanical stirrer, a reflux condenser and the mixture was heated at reflux for 8 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry. The cake was charged to a 5 L 3-neck flask and 1.6 L of MeOH was added. The flask was equipped with a mechanical stirrer, a water condenser and the contents were heated at reflux for 6 hours. After cooling overnight, the mixture was filtered through sharkskin filter paper and sucked dry.
The cake was dissolved into 1.6 L of HOAc with the assistance of gentle heating in the water bath of a rotary evaporator. The solution was filtered through #3 filter paper and the total volume of the filtrate was reduced to ~500 mL in volume on the rotary evaporator at 60 °C/60 mmHg. At this stage, the bulk of the mixture remained a yellow solution and a small amount of precipitate formed. To the flask was charged 500 mL of xylenes (precipitate formed) and the total volume was reduced to -500 mL in volume on the rotary evaporator at 60°C/60 mmHg. The process was repeated two more times. After cooling, the mixture was filtered, the cake was washed with 500 mL of xylenes and sucked dry. The cake was transferred to a glass dish and further dried at 80°C/27 inch vacuum overnight.
The cake was off-white in color and weighed 108.38g. X-ray powder diffraction analysis indicated that a crystalline form was present, which was characterized as Form IV by a powder X- ray diffraction pattern comprising peaks at the following approximate diffraction angles (20): 8.9, 12.0, 14.6, 15.2, 15.7, 17.8, 19.2, 20.5, 21.6, 23.2, 24.2, 24.8, 26.2, and 27.5.
While the invention has been illustrated by reference to specific and preferred embodiments, those skilled in the art will recognize that variations and modifications may be made through routine experimentation and practice of the invention. Thus, the invention is intended not to be limited by the foregoing description, but to be defined by the appended claims and their equivalents.
.............................
Chekal, B. P.; Guinness, S. M.; Lillie, B. M.; McLaughlin, R. W.; Palmer, C. W.; Post, R. J.; Sieser, J. E.; Singer, R. A.; Sluggett, G. W.; Vaidyanathan, R.; Withbroe, G. Org. Process Res. Dev. 2014, 18, 266 http://pubs.acs.org/doi/abs/10.1021/op400088k

Babu, S.; Dagnino, R., Jr.; Ouellette, M. A.; Shi, B.; Tian, Q.; Zook, S. E. PCT Int. Appl. WO/2006/048745, 2006.
.......................

....................................
http://www.google.com/patents/CN103570696A?cl=en

formula:

(1) 6-nitro-indazole dissolved in an aprotic solvent, and 3,4-dihydro -2H- pyran catalyst, 6-nitro-indazole in the catalyst and the 3,4-dihydro -2H - pyran reaction, the protecting group NH-position, was prepared to give 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:

Wherein the 3,4-dihydro -2H- pyran an amount of 3 equivalents wide;
Aprotic solvent is acetonitrile, ethyl acetate, toluene or xylene;
The catalyst is 2,3-dichloro-5,6-dicyano-p-benzoquinone, p-toluenesulfonic acid or methanesulfonic acid;
The reaction temperature is 7 (T90 ° C, the reaction time is 1 to 4 hours;
(2) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole dissolved in a polar aprotic solvent, iodine was added and the acid-binding agent, an inorganic base, to afford 3- iodo-6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (I), the reaction equation is:

Inorganic base acid binding agent is potassium carbonate, sodium carbonate, potassium hydroxide, sodium hydroxide, potassium bicarbonate, sodium bicarbonate, cesium carbonate, lithium hydroxide;
The reaction temperature is 2 (T40 ° C, the reaction time is 8 to 20 hours.
[0009] A Axitinib intermediate (1) in preparation for the Nepalese Asif application, based on intermediate (1) and 2-vinyl pyridine Heck coupling reaction, followed sequentially nitro reduction and the diazotization reaction of iodine, and finally with a 2-mercapto--N- methylbenzamide was prepared by deprotection docking axitinib, including the following synthetic steps:
(I) Intermediate (1) and be given 2_ vinylpyridine Jie Heck coupling reaction to give (E) _6_ nitro _3- [2_ (P than-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, the reaction equation is:





A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (I) 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added acetonitrile (2L), followed by addition of 6-nitro-indazole (163.1g, 1.0mol), 3, 4- dihydro -2H- pyran (168.2g, 2.0mol), 2,3- dichloro-5,6-dicyano-p-benzoquinone (22.7g, 0.1mol), was heated to 820C under reflux for 2 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirring I hour, delamination, the organic phase washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol, 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 236.3 g, yield 95.6%, m.p. 110 ~ 120 ° C, 1Η NMR (CDCl3): δ 1.30-1.83 (m, 6Η, Η3, _Η5,), 3.82-3.93 (m, 2Η, Η6 '), 5.86 (m , 1Η, Η2 '), 8.10-8.12 (m, 2Η, Η3, Η5), 8.31 (m, 1Η; Η4), 8.55 (s, 1Η, Η7);
The reaction equation is as follows:

5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, l.0eq) and potassium carbonate ( 251.6g, 1.82mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours , the reaction system was stirred at 25 ° C for 16 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature 30 ° C Hereinafter, stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid (Ι), 326.5g, yield 96.2%, m.p. 135 ~ 137 ° C / H NMR (DMS0_d6): δ 1.60-1.61 (m, 2H, H4,, H5 '), 1.73-1.76 (m, 1H, H5'), 2.01-2.04 (m, 2H, H3 ', H4'), 2.35-2.38 (m, 1H, H3 '), 3.81-3.87 (m, 2H, H6'), 6.11-6.14 (dd, 1H, H2 '), 7.70-7.72 (d , 1H, H4),
8.05-8.07 (dd, 1H, H5), 8.79 (s, 1H, H7).
The reaction equation is as follows:

Synthesis of (I) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin-2 - yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ', H5'), 1.79-1.81 (m, 1H, H5 '), 2.05-2.07 (m, 2H, H3', H4 '), 2.44-2.50 (m, 1H , H3 '), 3.86-3.90 (m, 2H, H6'), 6.15-6.18 (dd, 1H, H2 '), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ', H5 '), 2.02-2.06 (m, 1H, H5'), 2.17-2.18 (m, 1H, H4 '), 2.55-2.60 (m, 1H, H3') 3.70-3.72 (m, 2H, H3 ', H6 '), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6'), 5.57-5.60 (dd, 1H, H2 '), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2x1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ', H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

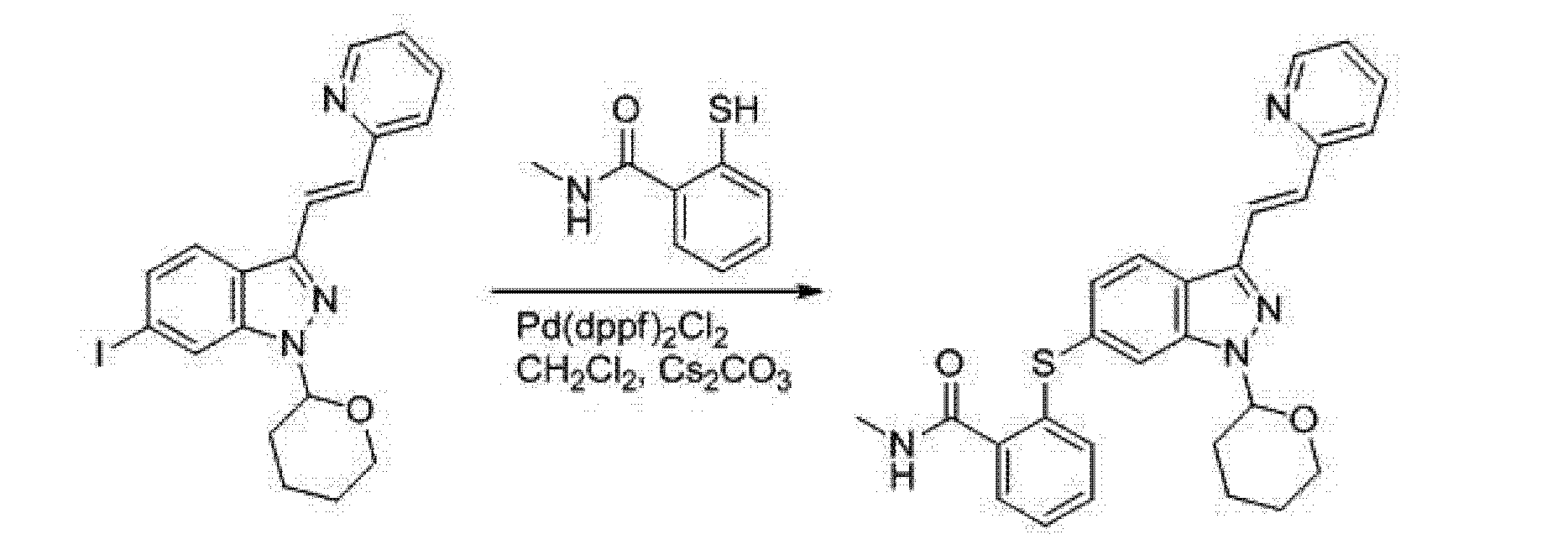
A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, - bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

In a 2L reaction flask was added (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H - indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added ethyl acetate (2L), followed by addition of 6-nitro-indazole (163.14g, 1.0mol), 3, 4- dihydro -2H- pyran (210.3g, 2.5mol), toluene acid (20.7g, 0.12mol), heated to 78 ° C under reflux for 3 hours to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile, recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid 223.3g, 6- nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, yield 90.3%, m.p. 110 ^ 11 TC;
The reaction equation is as follows:

5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.81mol, l.0eq) and sodium hydroxide (64.7g, 1.62mol, 2.0eq), ice-cooled (10 ° C or less), followed by stirring, iodine (369.6g, 1.46mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system, addition time 2 hours, the reaction system was stirred at 25 ° C for 12 hours to complete the reaction, sodium thiosulfate was added (198.2g, 1.25mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the temperature of 30 ° C or less, and stirred for 30 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 30 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 294.3g, 97.5% yield, m.p. 136 ~ 137. . .
[0014] The reaction equation is as follows:

Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2Η-) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ', H5'), 1.79-1.81 (m, 1H, H5 '), 2.05-2.07 (m, 2H, H3', H4 '), 2.44-2.50 (m, 1H , H3 '), 3.86-3.90 (m, 2H, H6'), 6.15-6.18 (dd, 1H, H2 '), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ', H5 '), 2.02-2.06 (m, 1H, H5'), 2.17-2.18 (m, 1H, H4 '), 2.55-2.60 (m, 1H, H3') 3.70-3.72 (m, 2H, H3 ', H6 '), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6'), 5.57-5.60 (dd, 1H, H2 '), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g, 0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I h, a solution process maintain an internal temperature of 0 ° C, the same temperature for I hour, dropping HCl solution (concentrated hydrochloric acid 112mL, water 200mL) at O ° C, the dropping time of 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). dropwise 800mL dichloromethane between 0 ° C, the dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) was dissolved water 600mL, in (TC dropwise added to the system at the same temperature for 2 hours to complete the reaction. The reaction mixture was poured into the system to 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layered , the aqueous phase was extracted with dichloromethane frozen (2x800mL), dichloromethane phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes , separated and the aqueous phase was extracted with dichloromethane (2x1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl ) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58- 1.61 (m, 2H, H4 ', H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,) , 3.79-3.81 (m, 1H, H6,), 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,), 7.29-7.31 (m, 1H, pyridine H5), 7.56 -7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4 ), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7), 8.61-8.62 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, - bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:

In a 2L reaction flask was added (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H - indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II), yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

A Assi intermediates for preparing Nigeria, comprising the steps of:
Synthesis of (1) 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
5L reaction flask in toluene (2L), followed by addition of 6-nitro-indazole (163.lg, 1.0mol), 3,4- dihydro -2H- pyran (193.5g, 2.3mol), methanesulfonic acid (14.4g, 0.15mol), heated to 85 ° C under reflux for 3.5 hours, to complete the reaction, cooled to room temperature, rotary evaporated to dryness, added water and dichloromethane 2L 2L, stirred for I hour, stratification, the organic phase was washed with brine wash, dried over anhydrous sodium sulfate, filtered, and rotary evaporated to dryness, and then dissolved in acetonitrile and 2L, stirring ice-salt bath chilled to _5 ° C for 2 hours, suction filtered, the filter cake washed with a small amount of cold acetonitrile and paint, and recrystallized from ethanol , 60 ° C and dried in vacuo 12 hours to give an off-white solid, 6-nitro-1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 234.4g, 94.8% yield, m.p. 111 ~ 112.. ;
The reaction equation is as follows:

5L reaction flask in DMF 700mL, followed by addition of 6-nitro-_1_ (tetrahydro -2H- pyran-2-yl) -1H- indazole (225.0g, 0.91mol, 1.0eq) and potassium hydroxide ( 102.lg, 1.82mol, 2.0eq), ice-cooled below 10 ° C, with stirring, iodine (415.8g, 1.64mol, 1.8eq) was dissolved in DMF 300mL, was added dropwise to the reaction system dropwise over 2 hours, The reaction system was stirred at 30 ° C for 10 hours to complete the reaction, sodium thiosulfate was added (223.0g, 1.41mol, 1.55eq) and 1.50g of potassium carbonate aqueous solution (1.5L), while maintaining the internal temperature below 30 ° C , stirred for 45 minutes at room temperature, water was added with stirring 2L, solid precipitated, stirred for 45 minutes at room temperature, suction filtered, the filter cake was washed with water, 60 ° C and dried in vacuo 12 hours to give a pale yellow solid
(1), 317.2g, 93.4% yield, m.p. 135 ~ 136 ° C.
The reaction equation is as follows:

Synthesis (1) (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole
A 5L reaction flask was added DMF (2L), followed by addition of the intermediate (1) (312.0g, 0.84mol), 2- vinylpyridine (127.5g, 1.21mol), N, N- diisopropylethylamine ( 205.3g, 1.59mol), tri-o-tolylphosphine (22.3g, 0.073mol) and palladium chloride (4.9g, 0.028mol), nitrogen, and heated to 100 ° C for 12 hours to complete the reaction, cooled to 45 ° C, isopropanol was added 1L, stirring at 45 ° C for 30 minutes, diluted with water and 5L, stirring at room temperature for I h, suction filtered, washed with water, isopropanol was added to the filter cake 1.2L, stirred at 55 ° C for 30 minutes, then stirred at room temperature for 30 minutes, suction filtered, the filter cake washed with cold isopropanol, 50 ° C and dried under vacuum for 12 hours to give (E) -6- nitro-3- [2- (pyridin _2 _-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole, 275.3g, 94.0% yield, m.p. 175 ~ 176 ^, ¾ NMR (DMSO-Cl6): δ 1.63-1.64 (m, 2H, H4 ', H5'), 1.79-1.81 (m, 1H, H5 '), 2.05-2.07 (m, 2H, H3', H4 '), 2.44-2.50 (m, 1H , H3 '), 3.86-3.90 (m, 2H, H6'), 6.15-6.18 (dd, 1H, H2 '), 7.30-7.33 (dd, 1H, pyridine H5), 7.65-7.69 (d, 1H, J = 16Hz, vinyl H2), 7.72-7.74 (d, 1H, pyridine H4), 7.82-7.86 (m, 1H, pyridine H3), 7.96-8.00 (d, 1H, J = 16Hz, vinyl HI), 8.07 -8.10 (dd, 1H, H4), 8.44-8.46 (d, 1H, H5), 8.63-8.64 (d, 1H, pyridine H6), 8.77-8.78 (d, 1H, H7);
The reaction equation is as follows:

5L reaction flask in ethanol HOOmLdjC 1000mL and ammonium chloride (300.0g, 5.61mol), was dissolved with stirring, followed by addition of (E) -6- nitro-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (255.0g, 0.73mol), was added iron powder (162.6g, 2.91mol), heated to 50 ° C the reaction was stirred for 2 hours to completion of the reaction, was cooled to 22 ° C, tetrahydrofuran 2L, stirred for I hour at room temperature, filtered through Celite, the filter cake washed with tetrahydrofuran and the filtrate was rotary evaporated to dryness, cooled to room temperature, water was added 2L, stirred for I hour at room temperature, pumping filtered, the filter cake washed with petroleum ether, 50 ° C and dried under vacuum for 12 hours to give a pale yellow solid 206.5g, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1Η- indazole, yield 88.6%, m.p. 162 ~ 164 ° C / H NMR (CDCl3): δ 1.63-1.77 (m, 2H, H4 ', H5 '), 2.02-2.06 (m, 1H, H5'), 2.17-2.18 (m, 1H, H4 '), 2.55-2.60 (m, 1H, H3') 3.70-3.72 (m, 2H, H3 ', H6 '), 3.91 (s, 2H, NH2), 4.04-4.07 (m, 1H, H6'), 5.57-5.60 (dd, 1H, H2 '), 6.64-6.66 (dd, 1H, H5), 6.74-6.75 (d, 1H, H7), 7.13-7.16 (dd, 1H, pyridine H5), 7.48-7.50 (d, 1H, pyridine H4), 7.49-7.53 (d, 1H, J = 16Hz, vinyl H2), 7.64 -7.68 (m, 1H, pyridine H3), 7.78-7.82 (d, 1H, J = 16Hz, vinyl Hl), 7.82-7.83 (d, 1H, H4), 8.60-8.61 (d, 1H, pyridine H6) ;
The reaction equation is as follows:

A 5L reaction flask was added 600mL of water and sodium nitrite (70.2g, 1.02mol), stirred and dissolved, and cooled to (TC, (E) -6- amino-3- [2- (pyridin-2-yl) ethenyl ] -1- (tetrahydro -2H- pyran-2-yl) -1H- indazole (200.0g,
0.62mol) was dissolved in glacial acetic acid 1.3L, dropwise added to the system dropwise over I hour, added dropwise to maintain the internal temperature process 0 ° C, the same temperature for I h, HCl solution was added dropwise at O ° C (112mL of concentrated hydrochloric acid , water 200mL), was added dropwise for 10 minutes, with the temperature for I h, TLC plate tracking point diazonium salt formation reaction (PE: EA = 1: 1). Solution of methylene chloride at 0 ° C and 800mL, dropping time of 5 minutes, potassium iodide (207.3g, l.25mol) and iodine (79.2g, 0.31mol) dissolved in water 600mL, at (TC dropwise added to the system, same temperature for 2 hours to complete the reaction. The reaction system was poured into a mixture of 20% sodium thiosulfate solution (2L) and dichloromethane SOOmL and stirred, layers were separated, the aqueous phase was extracted with dichloromethane frozen (2x800mL ), methylene chloride phases were combined burning, 3M sodium hydroxide solution was added dropwise 3.5L, adjust the aqueous phase pH = 9 ~ 12, and water was added ammonia 200mL 400mL, stirred for 30 minutes, separated and the aqueous phase extracted with dichloromethane ( 2x1.2L), the organic phases were combined, rotary evaporated to dryness, and purified through silica gel to give (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro -2H - pyran-2-yl) -1H- indazole, 176.0g, 65.4% yield, m.p. 142 ~ 143 ° C, 1H NMR (DMS0_d6): δ 1.58-1.61 (m, 2H, H4 ', H5,) 1.72-1.78 (m, 1H, H5,), 1.97-2.04 (m, 2H, H3,, H4,), 2.38-2.44 (m, 1H, H3,), 3.79-3.81 (m, 1H, H6,) , 3.88-3.90 (m, 1H, H6,), 5.91-5.94 (dd, 1H, H2,),
7.29-7.31 (m, 1H, pyridine H5), 7.56-7.60 (d, 1H ,, J = 16Hz, vinyl H2), 7.57-7.59 (m, 1H, pyridine H4), 7.69-7.71 (d, 1H, pyridine H3), 7.80-7.84 (m, 1H, H4), 7.89-7.93 (d, 1H, J = 16Hz, vinyl HI), 8.01-8.03 (d, 1H, H5), 8.25 (s, 1H, H7 ), 8.61-8.62 (d, 1H, pyridine H6); reaction equation is as follows:

A 5L reaction flask was added DMF (1750mL) and (E) -6- iodo-3- [2- (pyridin-2-yl) ethenyl] -1- (tetrahydro-pyran-2-yl -2H-) -1H- indazole (175.0g, 0.41mol), nitrogen, was added [1, I, - bis (diphenylphosphino) ferrocene] dichloropalladium dichloromethane complex (14.9g, 0.018mmol ), cesium carbonate (198.3g, 0.61mol) and dichloromethane 20mL, was added 2-mercapto -N- methylbenzamide (84.9g, 0.5Imol), heated to 80 ° C for 16 hours to complete the reaction, spin distilled was removed DMF, cooled to room temperature, ethyl acetate was added 3L, water 4L, stirred for 40 minutes, the organic phase was separated, washed with brine, layered, dried over sodium sulfate, filtered, and rotary evaporated to dryness, to give (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) -1- (tetrahydro -2H- pyran-2-yl) -1H- indazol-6-yl] thio } benzamide, 165.6g, a yield of 86.7%, the melting point of 142 ~ 143 ° C;
The reaction equation is as follows:
In a 2L reaction flask was added (E) -N- methyl-2 - {[3- (2- (pyridin-2-yl) ethenyl) _1_ (tetrahydro -2H- pyran-2-yl) -1H - indazol-6-yl] thio} benzamide (150.0g, 0.32mol), p-toluenesulfonic acid monohydrate (303.2g, 1.59mol), methanol (800mL) and water (150mL), nitrogen, heated to 65 ° C for 4 hours, spin evaporated to dryness and ethanol (800mL), 65 ° C was stirred for I hour, the ethanol was removed by rotary evaporation, then repeated three times, TLC spot plate tracking reaction (petroleum ether: ethyl acetate = 1: 1). Completion of the reaction, cooled to room temperature, rotary evaporated to dryness, water was added 500mL, stirred for I h, filtered, and the filter cake was washed with methanol and ice, and then added to the reaction vessel, ethyl acetate was added 450mL, stirred at 65 ° C 30 minutes. cooled to room temperature, suction filtered, the filter cake washed with ethyl acetate and freeze paint, water paint, 50 ° C and dried under vacuum for 12 hours to give a white solid 117.5g, Axitinib (II),
yield 95.4%, HPLC purity 98.8 % / H NMR (DMS0_d6): δ 2.78 (d, 3H, CH3), 7.05 (dd, 1H), 7.19 (dd, 1H), 7.36-7.23 (m, 3H), 7.50 (dd, 1H), 7.58 ( d, 1H), 7.61 (s, 1H), 7.66 (d, 1H), 7.85-7.76 (m, 1H), 7.96 (d, 1H, J = 16Hz), 8.21 (d, 1H), 8.39 (q, 1H), 8.61 (d, 1H), 13.35 (s, 1H).
The reaction equation is as follows:

.........................

...........................

...........
NMR source apexbt


http://dmd.aspetjournals.org/content/suppl/2014/03/07/dmd.113.056531.DC1/Supplemental__Data_Figures_56531.pdf
NMR..SEE........http://www.abmole.com/download/axitinib-hnmr.pdf

MASS






References
- "Inlyta (axitinib) dosing, indications, interactions, adverse effects, and more". Medscape Reference. WebMD. Retrieved 25 January 2014.
- Wilmes, LJ; Pallavicini, MG; Fleming, LM; Gibbs, J; Wang, D; Li, KL; Partridge, SC; Henry, RG; Shalinsky, DR; Hu-Lowe, D; Park, JW; McShane, TM; Lu, Y; Brasch, RC; Hylton, NM (April 2007). "AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging". Magnetic Resonance Imaging 25 (3): 319–27. doi:10.1016/j.mri.2006.09.041. PMID 17371720.
- Rini, B; Rixe, O; Bukowski, R; Michaelson, MD; Wilding, G; Hudes, G; Bolte, O; Steinfeldt, H; Reich, SD; Motzer, R (June 2005). "AG-013736, a multi-target tyrosine kinase receptor inhibitor, demonstrates anti-tumor activity in a Phase 2 study of cytokine-refractory, metastatic renal cell cancer (RCC)". Journal of Clinical Oncology ASCO Annual Meeting Proceedings 23 (16S): 4509.
- Rugo, HS; Herbst, RS; Liu, G; Park, JW; Kies, MS; Steinfeldt, HM; Pithavala, YK; Reich, SD; Freddo, JL; Wilding, G (August 2005). "Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results"(PDF). Journal of Clinical Oncology 23 (24): 5474–83. doi:10.1200/JCO.2005.04.192.PMID 16027439.
- "FDA Approves Inlyta for Advanced Renal Cell Carcinoma". Drugs.com. January 27, 2012.
- John Fauber, Elbert Chu (Oct 27, 2014). "The Slippery Slope: Is a Surrogate Endpoint Evidence of Efficacy?". Milwaukee Journal Sentinel/MedPage Today.
- Spano, JP; Chodkiewicz, C; Maurel, J; Wong, R; Wasan, H; Barone, C; Létourneau, R; Bajetta, E; Pithavala, Y; Bycott, P; Trask, P; Liau, K; Ricart, AD; Kim, S; Rixe, O (June 2008). "Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: an open-label randomised phase II study". Lancet 371(9630): 2101–2108. doi:10.1016/S0140-6736(08)60661-3. PMID 18514303.
- "Pfizer pancreatic cancer drug fails, trial halted". Reuters. January 30, 2009.
- "Pfizer’s Phase III Trial in mRCC Turns Up Positive Results". 19 Nov 2010.
- "ODAC Unanimously Supports Axitinib for Renal Cell Carcinoma". 7 Dec 2011.
- "INLYTA (axitinib) tablet, film coated [Pfizer Laboratories Div Pfizer Inc]". DailyMed. Pfizer Laboratories Div Pfizer Inc. September 2013. Retrieved 25 January 2014.
- "Inlyta : EPAR - Product Information" (PDF). European Medicines Agency. Pfizer Ltd. 17 December 2013. Retrieved 25 January 2014.
- "Inlyta 1 mg 3mg, 5 mg & 7mg film-coated tablets - Summary of Product Characteristics (SPC)". electronic Medicines Compendium. Pfizer Limited. 5 December 2013. Retrieved25 January 2014.
- "PRODUCT INFORMATION INLYTA (axitinib)" (PDF). TGA eBusiness Services. Pfizer Australia Pty Ltd. 5 July 2013. Retrieved 25 January 2014.
- Tea Pemovska,Eric Johnson,Mika Kontro,Gretchen A. Repasky,Jeffrey Chen,Peter Wells,Ciarán N. Cronin,Michele McTigue,Olli Kallioniemi,Kimmo Porkka,Brion W. Murray & Krister Wennerberg. "Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation". Nature. doi:10.1038/nature14119.
- "FDA Prescribing Information" (PDF). 30 Jan 2012.
- Escudier, B; Gore, M. "Axitinib for the Management of Metastatic Renal Cell Carcinoma" (PDF). Drugs in R&d 11 (2): 113–126. doi:10.2165/11591240-000000000-00000. PMC 3585900. PMID 21679004.
- Zhang Y (Jan 2014). "Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways.". J Mol Med Rep 9 (1): 83–90.doi:10.3892/mmr.2013.1781. PMID 24213221.
- [1] http://www.cancer.gov/cancertopics/druginfo/axitinib[2] http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm289439.htm[3] Kosugi M, Shimizu T, T. Migita, Chemistry Letters , 1978 , pp 13-14.[4] Organic Process Research & Development 2008 , 12, 869? 876.[5] Furstner A. Chem. Commun ., 2008 , 2873? 2875.[6] Thorarensen A. , Synlett , 2010 , 2 pp 219 - 222.
[7] http://en.wikipedia.org/wiki/Heck_reaction - where you can find the reaction mechanism and many other useful information.
[8] Aoyama, T., Synthesis , 2004 , 8 pp 1183-1186.
http://www.cosmoscience.org/blog/wp-content/uploads/2014/08/GSluggett-Application-of-Topochemical-Principles-and-Solid-State-Photoreactivity.pdf
update
http://www.google.com/patents/US20140248347?cl=en
Axitinib is a potent and selective inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2 and 3. These receptors are implicated in pathologic angiogenesis, tumor growth, and metastatic progression of cancer. Axitinib has been shown to potently inhibit VEGF-mediated endothelial cell proliferation and survival. Clinical trials are currently on-going to study the use of axitinib for the treatment of various cancers, including liver cancer, melanoma, mesothelioma, non-small cell lung cancer, prostate cancer, renal cell carcinoma, soft tissue sarcomas and solid tumors. Inlyta® (axitinib) has been approved in the United States, Europe, Japan and other jurisdictions for the treatment of renal cell carcinoma.
Axitinib, as well as pharmaceutically acceptable salts thereof, is described in U.S. Pat. No. 6,534,524. Methods of making axitinib are described in U.S. Pat. Nos. 6,884,890 and 7,232,910, in U.S. Publication Nos. 2006-0091067 and 2007-0203196 and in International Publication No. WO 2006/048745. Dosage forms of axitinib are described in U.S. Publication No. 2004-0224988. Polymorphic forms and pharmaceutical compositions of axitinib are also described in U.S. Publication Nos. 2006-0094763, 2008-0274192 and 2010-0179329.
Crystalline Form IV of axitinib API was characterized by the solid state NMR spectrum shown in Figure 4. The 13C chemical shifts of crystalline Form IV of axitinib API are shown in Table 9.
Table 9
i aC Chemical Shifts
Relative Intensity
[ppm]
170.0 46 i aC Chemical Shifts
Relative Intensity
[ppm]
154.3 34
146.8 31
143.2 60
142.0 61
136.9 23
133.5 33
131 .9 48
129.5 88
126.2 80
121 .2 100
1 19.6 46
27.7 41
26.1 36
Crystalline Form IV of axitinib in drug product, which was prepared as provided in Example 8, as characterized by the solid state NMR spectrum shown in Figures 5 and 6. The 13C chemical shifts of crystalline Form IV of axitinib in drug product are shown in Table 10.
Table 10
 (a) Peak shoulder.
Form IV axitinib in the pharmaceutical composition of the present
invention may be identified by a solid state nuclear magnetic resonance
comprising any one or more of the following 13C chemical
shifts expressed in parts per million: 170.0 ± 0.2, 154.2 ± 0.2, 143.3 ±
0.2, 142.1 ± 0.2, 133.4 ± 0.2, 126.3 ± 0.2, 121 .3 ± 0.2 and 27.8 ±
0.2.
(a) Peak shoulder.
Form IV axitinib in the pharmaceutical composition of the present
invention may be identified by a solid state nuclear magnetic resonance
comprising any one or more of the following 13C chemical
shifts expressed in parts per million: 170.0 ± 0.2, 154.2 ± 0.2, 143.3 ±
0.2, 142.1 ± 0.2, 133.4 ± 0.2, 126.3 ± 0.2, 121 .3 ± 0.2 and 27.8 ±
0.2.
Crystalline Form XXV of axitinib in drug product, which was prepared as provided in Example 8, was characterized by the solid state NMR spectrum shown in Figures 7 and 8. The 13C chemical shifts of crystalline Form XXV of axitinib in drug product are shown in Table 1 1 .
Table 1 1
 (a) Peak shoulder
(a) Peak shoulder
Form XXV axitinib in the pharmaceutical composition of the present invention may be identified by a solid state nuclear magnetic resonance comprising any one or more of the following 13C chemical shifts expressed in parts per million: 167.4 ± 0.2, 157.7 ± 0.2, 144.9 ± 0.2, 140.9 ± 0.2, 129.7 ± 0.2, 128.8 ± 0.2, 127.3 ± 0.2, 123.7 ± 0.2, 120.5 ± 0.2, 1 16.5 ± 0.2 and 25.4 ± 0.2.
Crystalline Form XLI of axitinib in drug product, which was prepared as provided in Example 8, was characterized by the solid state NMR spectrum shown in Figures 9 and 10. The 13C chemical shifts of crystalline Form XLI of axitinib in drug product are shown in Table 12. Table 12
 Form XLI axitinib in the pharmaceutical composition
of the present invention may be identified by a solid state nuclear
magnetic resonance comprising any one or more of the following 13C
chemical shifts expressed in parts per million: 142.6 ± 0.2, 136.8 ±
0.2, 136.2 ± 0.2, 133.7 ± 0.2, 132.1 ± 0.2, 121 .4 ± 0.2 and 1 19.8 ±
0.2.
Form XLI axitinib in the pharmaceutical composition
of the present invention may be identified by a solid state nuclear
magnetic resonance comprising any one or more of the following 13C
chemical shifts expressed in parts per million: 142.6 ± 0.2, 136.8 ±
0.2, 136.2 ± 0.2, 133.7 ± 0.2, 132.1 ± 0.2, 121 .4 ± 0.2 and 1 19.8 ±
0.2.
Polymorphic Form IV of axitinib API and polymorphic Forms IV, XXV, and XLI, of axitinib within the drug product or pharmaceutical composition of the present invention were each characterized using Raman spectroscopy. The Raman spectra differ for each of the polymorphic forms of formulated axitinib. For example, Forms IV, XXV, and XLI of axitinib in drug product can be distinguished from each other and from other polymorphic forms of formulated axitinib by using Raman spectroscopy. The detection of characteristic Raman spectra of axitinib within the drug product or pharmaceutical composition of the present invention enables unique identification of polymorphic Forms IV, XXV, and XLI, of axitinib in the drug product or pharmaceutical composition.
Raman spectra of Form IV of axitinib API were collected using a Nicolet NXR FT- Raman accessory attached to a Nicolet 6700 FTIR spectrometer equipped with a KBr beamsplitter and a d-TGS KBr detector. The spectrometer is equipped with a 1064 nm Nd:YVO4 laser and a liquid nitrogen cooled Germanium detector. Prior to data acquisition, instrument performance and calibration verifications were conducted using polystyrene. Samples were analyzed in glass NMR tubes that were spun during spectral collection. The spectra were collected using 0.5 W of laser power and 400 co- added scans. The collection range was 3700-300 cm"1. The API spectra were recorded using 2 cm"1 resolution, and Happ-Genzel apodization was utilized for all of the spectra. A single spectrum was recorded for each sample, which was intensity normalized prior to peak picking.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
 LIONEL MY SON
LIONEL MY SON

update
http://www.google.com/patents/US20140248347?cl=en
Axitinib is a potent and selective inhibitor of vascular endothelial growth factor (VEGF) receptors 1, 2 and 3. These receptors are implicated in pathologic angiogenesis, tumor growth, and metastatic progression of cancer. Axitinib has been shown to potently inhibit VEGF-mediated endothelial cell proliferation and survival. Clinical trials are currently on-going to study the use of axitinib for the treatment of various cancers, including liver cancer, melanoma, mesothelioma, non-small cell lung cancer, prostate cancer, renal cell carcinoma, soft tissue sarcomas and solid tumors. Inlyta® (axitinib) has been approved in the United States, Europe, Japan and other jurisdictions for the treatment of renal cell carcinoma.
Axitinib, as well as pharmaceutically acceptable salts thereof, is described in U.S. Pat. No. 6,534,524. Methods of making axitinib are described in U.S. Pat. Nos. 6,884,890 and 7,232,910, in U.S. Publication Nos. 2006-0091067 and 2007-0203196 and in International Publication No. WO 2006/048745. Dosage forms of axitinib are described in U.S. Publication No. 2004-0224988. Polymorphic forms and pharmaceutical compositions of axitinib are also described in U.S. Publication Nos. 2006-0094763, 2008-0274192 and 2010-0179329.
Crystalline Form IV of axitinib API was characterized by the solid state NMR spectrum shown in Figure 4. The 13C chemical shifts of crystalline Form IV of axitinib API are shown in Table 9.
Table 9
i aC Chemical Shifts
Relative Intensity
[ppm]
170.0 46 i aC Chemical Shifts
Relative Intensity
[ppm]
154.3 34
146.8 31
143.2 60
142.0 61
136.9 23
133.5 33
131 .9 48
129.5 88
126.2 80
121 .2 100
1 19.6 46
27.7 41
26.1 36
Crystalline Form IV of axitinib in drug product, which was prepared as provided in Example 8, as characterized by the solid state NMR spectrum shown in Figures 5 and 6. The 13C chemical shifts of crystalline Form IV of axitinib in drug product are shown in Table 10.
Table 10

Crystalline Form XXV of axitinib in drug product, which was prepared as provided in Example 8, was characterized by the solid state NMR spectrum shown in Figures 7 and 8. The 13C chemical shifts of crystalline Form XXV of axitinib in drug product are shown in Table 1 1 .
Table 1 1

Form XXV axitinib in the pharmaceutical composition of the present invention may be identified by a solid state nuclear magnetic resonance comprising any one or more of the following 13C chemical shifts expressed in parts per million: 167.4 ± 0.2, 157.7 ± 0.2, 144.9 ± 0.2, 140.9 ± 0.2, 129.7 ± 0.2, 128.8 ± 0.2, 127.3 ± 0.2, 123.7 ± 0.2, 120.5 ± 0.2, 1 16.5 ± 0.2 and 25.4 ± 0.2.
Crystalline Form XLI of axitinib in drug product, which was prepared as provided in Example 8, was characterized by the solid state NMR spectrum shown in Figures 9 and 10. The 13C chemical shifts of crystalline Form XLI of axitinib in drug product are shown in Table 12. Table 12

Polymorphic Form IV of axitinib API and polymorphic Forms IV, XXV, and XLI, of axitinib within the drug product or pharmaceutical composition of the present invention were each characterized using Raman spectroscopy. The Raman spectra differ for each of the polymorphic forms of formulated axitinib. For example, Forms IV, XXV, and XLI of axitinib in drug product can be distinguished from each other and from other polymorphic forms of formulated axitinib by using Raman spectroscopy. The detection of characteristic Raman spectra of axitinib within the drug product or pharmaceutical composition of the present invention enables unique identification of polymorphic Forms IV, XXV, and XLI, of axitinib in the drug product or pharmaceutical composition.
Raman spectra of Form IV of axitinib API were collected using a Nicolet NXR FT- Raman accessory attached to a Nicolet 6700 FTIR spectrometer equipped with a KBr beamsplitter and a d-TGS KBr detector. The spectrometer is equipped with a 1064 nm Nd:YVO4 laser and a liquid nitrogen cooled Germanium detector. Prior to data acquisition, instrument performance and calibration verifications were conducted using polystyrene. Samples were analyzed in glass NMR tubes that were spun during spectral collection. The spectra were collected using 0.5 W of laser power and 400 co- added scans. The collection range was 3700-300 cm"1. The API spectra were recorded using 2 cm"1 resolution, and Happ-Genzel apodization was utilized for all of the spectra. A single spectrum was recorded for each sample, which was intensity normalized prior to peak picking.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
TAKE A TOUR

Palau Islands
Geographically Located in Micronesia, 7° 30' North Latitude, 133° 30' East Longitude, Palau is an island nation comprised of 16 states. Officially the Republic of Palau, this island nation in the Pacific Ocean is about 500 miles east of the Philippines and 2000 miles south of Tokyo.

Climate: Palau enjoys a pleasantly warm climate all year round with an annual mean temperature of 82° degrees F. (27° C.). Rainfall can occur throughout the year, and the annual average is 150 inches. The average relative humidity is 82%, and although rain falls more frequently between July and October, there is still much sunshine. Typhoons are rare as Palau is located outside the typhoon zone.


Getting There: From the western seaboard of the United States, you can hop to Hawaii, skip to Guam, then jump to Palau. For a scenic island route, you can do an island hop across Micronesia to Palau. Through Asia, there are twice weekly charter services between Taipei, Taiwan and Palau and additional flights are also available during peak seasons. From Europe,visitors can fly via Emerates direct to Manila, Philippines and onwards with Continental Airlines to Palau--this is possible without overnight stay in Manila.
Getting There: Australia, Fly from any major cities in Australia to Cairns to make your connection to Palau. Continental Airlines flies between Cairns and Guam twice weekly and daily from Guam to Palau. Most carriers service Manila, and connect via Continental Airlines’ twice-weekly flights to Palau






Palau - Wikipedia, the free encyclopedia
https://en.wikipedia.org/wiki/Palau




Fruit Bat Soup - a delicacy on the island of Palau | The world's ...

Palau Style Broiled Fish Recipe | Island Food | Pinterest
www.pinterest.com674 × 402Search by image
///////
Palau Style Broiled Fish RecipePalau Style, Amazing Recipes, Food Fish, Style Broil,