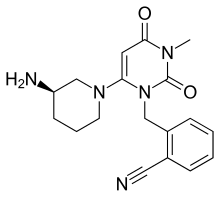
ALOGLIPTIN
Alogliptin is a potent, selective inhibitor of DPP-4 with IC50 of <10 nM, exhibits greater than 10,000-fold selectivity over DPP-8 and DPP-9.
Alogliptin (trade name Nesina in the US[1] and Vipidia in Europe[2]) is an orally administered anti-diabetic drug in the DPP-4 inhibitor class,[3] developed by Syrrx, a company which was acquired by Takeda Pharmaceutical Company in 2005. Like other medications for the treatment of Type 2 diabetes, alogliptin does not decrease the risk of heart attack and stroke. Like other members of the gliptin class, it causes little or no weight gain, exhibits relatively little risk of causing hypoglycemia, and exhibits relatively modest glucose-lowering activity. Alogliptin and other gliptins are commonly used in combination with metformin in patients whose diabetes cannot adequately be controlled with metformin alone.[4]
Clinical study
Alogliptin is a dipeptidyl peptidase-4 inhibitor (DPP-4i) that is designed to slow the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide). [5] A randomized clinical trial reporting in 2011 aimed to determine the efficacy and safety of alogliptin versus placebo and vogliboseamong newly diagnosed Type 2 diabetes patients in Japan. The main outcome indicated that alogliptin was statistically superior to both comparitors.[6] A randomized clinical trial reporting in 2012 aimed to demonstrate that alogliptin was "non-inferior" to a "very low fat/calorie traditional Japanese diet" among newly diagnosed Type 2 diabetes patients in Japan. The outcome indicated that both the drug and dietary treatments comparably impacted indicators of the diabetic condition, such as HbA1c levels and glycemic efficacy. The drug treatment had its impact without changing body mass index (BMI), but the dietary treatment was accompanied by a significant reduction in the BMI.[7] A randomized clinical trial reporting in 2011 aimed to demonstrate the efficacy of alogliptin as an add-on agent in combination withmetformin and pioglitazone versus simply increasing the dosage of pioglitazone in combination with metformin; in other words, this was a study to look at a three-agent therapy versus a two-agent therapy. The outcome of this study suggested that the addition of alogliptin to metformin and pioglitazone provided superior impact on diabetes biomarkers (e.g. HbA1c) than increasing the dose of pioglitazone in a two agent therapy with metformin.[8]Reported adverse events
Adverse events appear to be restricted to mild hypoglycemia based on clinical studies.[6][7][8] Alogliptin is not associated with increased weight, increased risk of cardiovasular events, or heart failure.[9][10]Market access
In December 2007, Takeda submitted a New Drug Application (NDA) for alogliptin to the United States Food and Drug Adminiistration (USFDA),[11] after positive results from Phase III clinical trials.[1] In September of 2008, the company also filed for approval in Japan,[12] winning approval in April 2010.[11] The company also filed a Marketing Authorization Application (MAA) elsewhere outside the United States, which was withdrawn in June 2009 needing more data.[12] The first USFDA NDA failed to gain approval and was followed by a pair of NDAs (one for alogliptin and a second for a combination of alogliptin and pioglitazone) in July 2011.[11] In 2012, Takeda received a negative response from the USFDA on both of these NDAs, citing a need for additional data.[11] In 2013 the FDA approved the drug in three formulations: As a stand-alone with the brand-name Nesina. Combined with metforminusing the name Kazano, and when combined with pioglitazone as Oseni. Diabetes affects millions of people worldwide and is considered one of the main threats to human health in the 21st century. In 2006, the World Health Organization (WHO) estimated that over 180 million people worldwide had diabetes, and the number is projected to double by 2030. Over time, uncontrolled diabetes can damage body systems, including the heart, blood vessels, eyes, kidneys and nerves. According to the WHO, approximately 1.1 million people died from diabetes in 2005, and it is estimated that diabetes-related deaths will increase by more than 50% in the next decade. Globally, the socioeconomic burden of diabetes is substantial. There are two main types of diabetes, designated type 1 and type 2, with type 2 diabetes accounting for over 90% of all diabetes cases globally. Type 1 diabetes is characterized by insulin deficiency, primarily caused by autoimmune-mediated destruction of pancreatic islet β-cells, and type 2 diabetes is characterized by abnormal insulin secretion and concomitant insulin resistance. To prevent the development of ketoacidosis, people with type 1 diabetes must take exogenous insulin for survival. Although those with type 2 diabetes are not dependent on exogenous insulin as much as subjects with type 1 diabetes, they may require exogenous insulin to control blood glucose levels.

.............
http://www.google.com/patents/EP2410855A1?cl=en
EXAMPLE 1 Preparation of (R)-2-((6-(3 -aminopiperidin-l-yl)-3 -methyl-2,4-dioxo-3 ,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-chloro-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 3):

EXAMPLE 2: Preparation of (R)-2-((6-(3-aminopiperidin-l-yl)-3-methyl-2,4-dioxo-3,4- dihydropyrimidin-1 (2H)-yl) methyl)benzonitrile (alogliptin) via 6-amino-l-(2- isocyanobenzyl)-3-methylpyrimidine-2,4(lH,3H)-dione (Scheme 4)


. .................. Patent EP2410855A1 http://www.google.com/patents/EP2410855A1?cl=en
 ..............
http://photo.blog.sina.com.cn/list/blogpic.php?pid=53891ebegd4e8671b28dc&bid=53891ebe0101grmv&uid=1401495230
..............
http://photo.blog.sina.com.cn/list/blogpic.php?pid=53891ebegd4e8671b28dc&bid=53891ebe0101grmv&uid=1401495230
 NMR
NMR
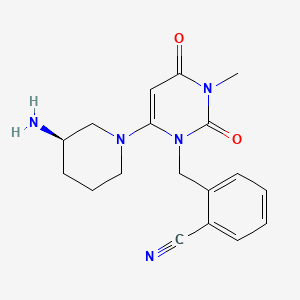 SOURCE APEXBT
SOURCE APEXBT

 References
References
- "Takeda Submits New Drug Application for Alogliptin (SYR-322) in the U.S." (Press release). Takeda Pharmaceutical Company. January 4, 2008. Retrieved January 9, 2008.
- Vipidia: EPAR summary for the public (European Medicines Agency)
- Feng, Jun; Zhang, Zhiyuan; Wallace, Michael B.; Stafford, Jeffrey A.; Kaldor, Stephen W.; Kassell, Daniel B.; Navre, Marc; Shi, Lihong; Skene, Robert J.; Asakawa, Tomoko; Takeuchi, Koji; Xu, Rongda; Webb, David R.; Gwaltney II, Stephen L. (2007). "Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV". J. Med. Chem.50 (10): 2297–2300.doi:10.1021/jm070104l.PMID 17441705.
- "www.aace.com" (PDF).
- http://www.takeda.com/news/2013/20130618_5841.html
- Seino, Yutaka; Fujita, Tetsuya; Hiroi, Shinzo; Hirayama, Masashi; Kaku, Kohei (September 2011), "Efficacy and safety of alogliptin in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, dose-ranging comparison with placebo, followed by a long-term extension study (abstract only)", Current Medical Research and Opinion 27 (9): 1781–1792,doi:10.1185/03007995.2011.599371,PMID 21806314, retrieved April 26,2012
- Kutoh, Eiji; Ukai, Yasuhiro (2012),"Alogliptin as an initial therapy in patients with newly diagnosed, drug naïve type 2 diabetes: a randomized, control trial (abstract only)", Endocrine(January 17, 2012), doi:10.1007/s12020-012-9596-0, PMID 22249941, retrieved April 26, 2012
- Bosi, Emanuele; Ellis, G.C.; Wilson, C.A.; Fleck, P.R. (October 2011), "Alogliptin as a third oral antidiabetic drug in patients with type 2 diabetes and inadequate glycaemic control on metformin and pioglitazone: a 52-week, randomized, double-blind, active-controlled, parallel-group study", Diabetes, Obesity and Metabolism (October 27, 2011) 13 (12): 1088–1096, doi:10.1111/j.1463-1326.2011.01463.x, retrieved April 26,2012
- White WB, Cannon CP, Heller SR et al. (October 2013). "Alogliptin after acute coronary syndrome in patients with type 2 diabetes". N. Engl. J. Med. 369(14): 1327–35.doi:10.1056/NEJMoa1305889.PMID 23992602.
- White WB, Zannad F (January 2014). "Saxagliptin, alogliptin, and cardiovascular outcomes". N. Engl. J. Med. 370 (5): 484.doi:10.1056/NEJMc1313880.PMID 24482824.
- Grogan, Kevin (April 26, 2012),"FDA wants yet more data on Takeda diabetes drug alogliptin",PharmaTimes (PharmaTimes), PharmaTimes online, retrieved April 26,2012
- "GEN News Highlights: Takeda Pulls MAA for Type 2 Diabetes Therapy". Genetic Engineering & Biotechnology News. June 4, 2009.
| EP1083172A1 * | May 26, 1998 | Mar 14, 2001 | Rimma Iliinichna Ashkinazi | N-substituted derivatives of 5-oxyiminobarbituric acid |
| US2598936 * | Apr 13, 1950 | Jun 3, 1952 | Searle & Co | Disubstituted cyanoalkanoylureas and thioureas and methods for their production |
| US6066641 * | Dec 12, 1995 | May 23, 2000 | Euro-Celtique S.A. | Aryl thioxanthines |
| US6248746 * | Jan 7, 1999 | Jun 19, 2001 | Euro-Celtique S.A. | 3-(arylalkyl) xanthines |
| US20080194593 * | Jan 11, 2008 | Aug 14, 2008 | Rao Kalla | A2b adenosine receptor antagonists |
| WO1994003456A1 * | Aug 5, 1993 | Feb 17, 1994 | Boehringer Ingelheim Kg | Asymmetrically substituted xanthine with adenosine-antagonistic properties |
| WO2001029010A1 * | Oct 18, 2000 | Apr 26, 2001 | Amjad Ali | Gram-positive selective antibacterial compounds, compositions containing such compounds and methods of treatment |
| WO2007035629A2 * | Sep 15, 2006 | Mar 29, 2007 | Takeda Pharmaceutical | Process for the preparation of pyrimidinedione derivatives |
| WO2007150011A2 * | Jun 22, 2007 | Dec 27, 2007 | Smithkline Beecham Corp | Prolyl hydroxylase inhibitors |
 |
|
| Systematic (IUPAC) name | |
|---|---|
2-({6-[(3R)-3-aminopiperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl}methyl)benzonitrile
|
|
| Clinical data | |
| Trade names | Nesina, Vipidia Kazano, Vipidomet (withmetformin) Oseni, Incresync (withpioglitazone) |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | 20% |
| Metabolism | Limited, hepatic (CYP2D6- and3A4-mediated) |
| Biological half-life | 12–21 hours |
| Excretion | Renal (major) and fecal (minor) |
| Identifiers | |
| CAS Registry Number | 850649-62-6 |
| ATC code | A10BH04 |
| PubChem | CID: 11450633 |
| IUPHAR/BPS | 6319 |
| ChemSpider | 9625485 |
| UNII | JHC049LO86 |
| KEGG | D06553 |
| ChEBI | CHEBI:72323 |
| ChEMBL | CHEMBL376359 |
| Synonyms | SYR-322 |
| Chemical data | |
| Formula | C18H21N5O2 |
| Molecular mass | 339.39 g/mol |
 Alogliptin benzoate
Alogliptin benzoate
| MF: | C18H21N5O2.C7H6O2 |
| MW: | 461.519 |
| Melting Point: | 185-188°C |
| Optical Rotation: | -56.3° (c=1, MeOH) |
 LIONEL MY SON
LIONEL MY SON

IMAGE...http://www.apexbt.com/
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
He
was only in first standard in school when I was hit by a deadly one in a
million spine stroke called acute transverse mylitis, it made me 90%
paralysed and bound to a wheel chair, Now I keep him as my source of
inspiration and helping millions, thanks to millions of my readers who
keep me going and help me to keep my son happy

TAKE A TOUR
TONGA
Kingdom of Tonga
Puleʻanga Fakatuʻi ʻo Tonga
 |  |
| Flag | Coat of arms |
Tonga - Wikipedia, the free encyclopedia
en.wikipedia.org/wiki/Tonga
Tonga ([ˈtoŋa]; Tongan: Puleʻanga Fakatuʻi ʻo Tonga), officially the Kingdom of Tonga, is a Polynesian sovereign state and archipelago comprising 177 ...
You may not have heard of Tonga, but if you are an adventure buff, then you are definitely in for a treat. Tonga comprises 150 islands, 36 of which are inhabited and lived by people. These islands are very small, but offer very big in terms of tourism and leisure. Also, the guys love to try the customs of Tonga.
Marriage Ceremonies
To maximize the thrill of the experience, make sure you attend at least one marriage ceremony in Tonga. This should not be too hard as Tonga are very helpful and will be more than willing to invite into their ceremonies and festivities. Marriage ceremonies in Tonga are sensational and they love to attend one.
Food Tonga
The way to eat your food Tonga is certainly very different from the way they eat yours. Given that, do not forget to try the local food in Tonga. One is the Lu Pulu - corned beef with a touch of coconut cream and cooked with taro leaves. There are also local beverages in Tonga, including kava, made from pepper plant.
Tonga Customs
The customs of Tonga are very native and unique. Here you will find that people are very friendly and welcoming. Make sure you try to assimilate their culture and customs are very appreciated in Tonga, and certainly do not want to forget this, not to offend a native of Tonga.
Sports
In Tonga, the most popular sport is rugby. Whether or not you are a rugby player, make sure you play this sport in Tonga. The experience will be very different from their country of origin, given the environment of Tonga in the rugby being played.
Beaches of Tonga
Logically, Tonga is home to a lot of beaches. By far, which means the islands of Tonga 150, at least half of the accessible beaches. Here, you can have activities such as sunbathing, playing or even a picnic with his family.
The lives of animals
Although animals are not in abundance in Tonga, are nevertheless given prime importance. Flying foxes and iguanas are everywhere. You can even treat them as exotic foods, however, this practice is discouraged.
Island Hopping
If you are an adventurous person and likes the idea of going from one island to the other, then Tonga is definitely the best place for you. The Tonga islands are very close to each other. The best thing to do is that you can take a boat from one island to another, allowing the excitement and fun of the Greek islands.
Fun water activities
In Tonga, it is fun. And what's more fun than surfing in the Pacific? The tides in Tonga are perfect for this kind of water fun you should try, and you can try water skiing or canoeing water in these places like this will be fun for you and your family.
Diving in Tonga
Another fun water activities you can try in Tonga is scuba diving. Now, why is diving in Tonga much different than elsewhere? Just because Tonga is located at the bottom of the Pacific Ocean, the sea is abundant wildlife in which you may see water species rare and stunning coral formations through their own eyes.
Mountain Activities
Volcanoes and mountains are in abundance in Tonga. This allows you to have a final trekking experience. In Tonga, such activities are managed in a very safe, and can also get to see volcanoes in a very close view.





Hopping Island





Tongan Food




Tongan Beaches
 PALACE
PALACE/////////