
CANAGLIFLOZIN
Canagliflozin
Canagliflozin is a highly potent and selective subtype 2 sodium-glucose transport protein (SGLT2) inhibitor to CHO- hSGLT2, CHO- rSGLT2 and CHO- mSGLT2 with IC50 of 4.4 nM, 3.7 nM and 2 nM, respectively.
M.F.C24H25FO5S
M.Wt: 444.52
CAS No: 842133-18-0
(1S)-1,5-Anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
TA 7284
JNJ 28431754
NMR.....http://file.selleckchem.com/downloads/nmr/S276003-Canagliflozin-HNMR-Selleck.pdf
| Canagliflozin Hemihydrate (1S)-1,5-Anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol hydrate (2:1) |  | 928672-86-0 |
(1S)-1,5-Anhydro-1-C-(3-{[5-(4-fluorophenyl)thiophen-2-yl]methyl}-4-methylphenyl)-d-glucitol hemihydrate
Molecular formula: C24H25FO5S,½H2O =453.5
CAS: 842133-18-0 (anhydrous
canagliflozin);
928672-86-0 (
canagliflozinhemihydrate)
Canagliflozin (INN, trade name Invokana) is a drug of the gliflozin class, used for the treatment of type 2 diabetes.[1][2] It was developed by Mitsubishi Tanabe Pharma and is marketed under license by Janssen, a division of Johnson & Johnson.[3]
U.S. Patent No, 7,943,788 B2 (the '788 patent) discloses canagliflozin or salts thereof and the process for its preparation.
U.S. Patent Nos. 7,943,582 B2 and 8,513,202 B2 discloses crystalline form of 1 -(P-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene hemihydrate and process for preparation thereof. The US '582 B2 and US '202 B2 further discloses that preparation of the crystalline form of hemi-hydrate canagliflozin typically involves dissolving in a good solvent (e.g. ketones or esters) crude or amorphous compound prepared in accordance with the procedures described in WO 2005/012326 pamphlet, and adding water and a poor solvent (e.g. alkanes or ethers) to the resulting solution, followed by filtration.
U.S. PG-Pub. No. 2013/0237487 Al (the US '487 Al) discloses amorphous dapagliflozin and amorphous canagliflozin. The US '487 Al also discloses 1:1 crystalline complex of canagliflozin with L-proline (Form CS1), ethanol solvate of a 1: 1 crystalline complex of canagliflozin with D-proline (Form CS2), 1 :1 crystalline complex of canagliflozin with L-phenylalanine (Form CS3), 1:1 crystalline complex of canagliflozin with D-proline (Form CS4).
The US '487 Al discloses preparation of amorphous canagliflozin by adding its heated toluene solution into n-heptane. After drying in vacuo the product was obtained as a white solid of with melting point of 54.7°C to 72.0°C. However, upon repetition of the said experiment, the obtained amorphous canagliflozin was having higher amount of residual solvents. Therefore, the amorphous canagliflozin obtained by process as disclosed in US '487 Al is not suitable for pharmaceutical preparations.
The US '487 Al further discloses that amorphous canagliflozin obtained by the above process is hygroscopic in nature which was confirmed by Dynamic vapor sorption (DVS) analysis. Further, it was observed that the amorphous form underwent a physical change between the sorption/desorption cycle, making the sorption/desorption behavior different between the two cycles. The physical change that occurred was determined to be a conversion or partial conversion from the amorphous state to a crystalline state. This change was supported by a change in the overall appearance of the sample as the humidity increased from 70% to 90% RH.
The canagliflozin assessment report EMA/718531/2013 published by EMEA discloses that Canagliflozin hemihydrate is a white to off-white powder^ practically insoluble in water and freely soluble in ethanol and non-hygroscopic. Polymorphism has been observed for canagliflozin and the manufactured Form I is a hemihydrate, and an unstable amorphous Form II. Form I is consistently produced by the proposed commercial synthesis process. Therefore, it is evident from the prior art that the reported amorphous form of canagliflozin is unstable and hygroscopic as well as not suitable for pharmaceutical preparations due to higher amount of residual solvents above the ICH acceptable limits.
Medical use
- Canagliflozin
is an antidiabetic drug used to improve glycemic control in people with
type 2 diabetes. In extensive clinical trials, canagliflozin produced a
consistent dose-dependent reduction in HbA1c
of 0.77% to 1.16% when administered as monotherapy, combination with
metformin, combination with metformin & Sulfonyulrea, combination
with metformin & pioglitazone and In combination with insulin from a
baselines of 7.8% to 8.1%, in combination with metformin, or in combination with metformin and a sulfonylurea.
When added to metformin Canagliflozin 100mg was shown to be
non-inferior to both Sitagliptin 100mg and glimiperide in reductions on
HbA1c at one year, whilst canagliflozin 300mg successfully demontrated
statistical superiority over both Sitagliptin and glimiperide in HbA1c
reductions. Secondary efficacy endpoint of superior body weight
reduction and blood pressure reduction (versus Sitagliptin and
glimiperide)) were observed as well. Canagliflozin produces beneficial
effects on HDL cholesterol whilst increasing LDL cholesterol to produce no change in total cholesterol.[4][5]
Contraindications
Canagliflozin has proven to be clinically effective in people with moderate renal failure and treatment can be continued in patients with renal impairment.
Adverse effects
Canagliflozin, as is common with all sglt2 inhibitors, increased (generally mild) urinary tract infections, genital fungal infections, thirst,[6] LDL cholesterol, and was associated with increased urination and episodes of low blood pressure.
There are concerns it may increase the risk of diabetic ketoacidosis.[7]
Cardiovascular problems have been discussed with this class of drugs.[citation needed] The pre-specified endpoint for cardiovascular safety in the canagliflozin clinical development program was Major Cardiovascular Events Plus (MACE-Plus), defined as the occurrence of any of the following events: cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, or unstable angina leading to hospitalization. This endpoint occurred in more people in the placebo group (20.5%) than in the canagliflozin treated group (18.9%).
Nonetheless, an FDA advisory committee expressed concern regarding the cardiovascular safety of canagliflozin. A greater number of cardiovascular events was observed during the first 30 days of treatment in canagliflozin treated people (0.45%) relative to placebo treated people (0.07%), suggesting an early period of enhanced cardiovascular risk. In addition, there was an increased risk of stroke in canagliflozin treated people. However none of these effects were seen as statistically significant. Additional cardiovascular safety data from the ongoing CANVAS trial is expected in 2015.[8]
Interactions
The drug may increase the risk of dehydration in combination with diuretic drugs.
Because it increases renal excretion of glucose, treatment with canagliflozin prevents renal reabsorption of 1,5-anhydroglucitol, leading to artifactual decreases in serum 1,5-anhydroglucitol; it can therefore interfere with the use of serum 1,5-anhydroglucitol (assay trade name, GlycoMark) as a measure of postprandial glucose excursions.[9]
Mechanism of action
Canagliflozin is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the renal glucose reabsorption (SGLT1 being responsible for the remaining 10%). Blocking this transporter causes up to 119 grams of blood glucose per day to be eliminated through the urine,[10] corresponding to 476 kilocalories. Additional water is eliminated by osmotic diuresis, resulting in a lowering of blood pressure.
This mechanism is associated with a low risk of hypoglycaemia (too low blood glucose) compared to other antidiabetic drugs such as sulfonylurea derivatives and insulin.[11]
History
On July 4, 2011, the European Medicines Agency approved a paediatric investigation plan and granted both a deferral and a waiver for canagliflozin (EMEA-001030-PIP01-10) in accordance with EC Regulation No.1901/2006 of the European Parliament and of the Council.[12]
In March 2013, canagliflozin became the first SGLT2 inhibitor to be approved in the United States.[13]
SYNTHESIS
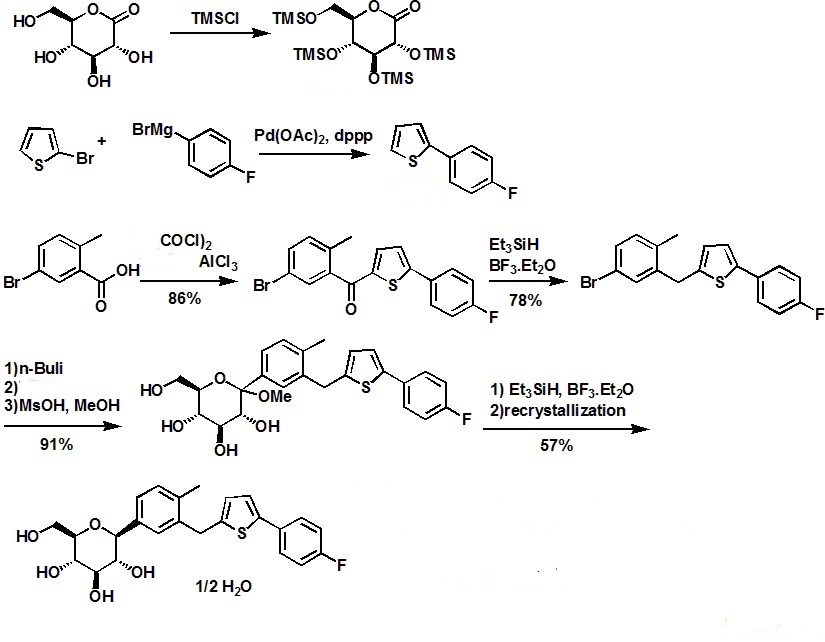
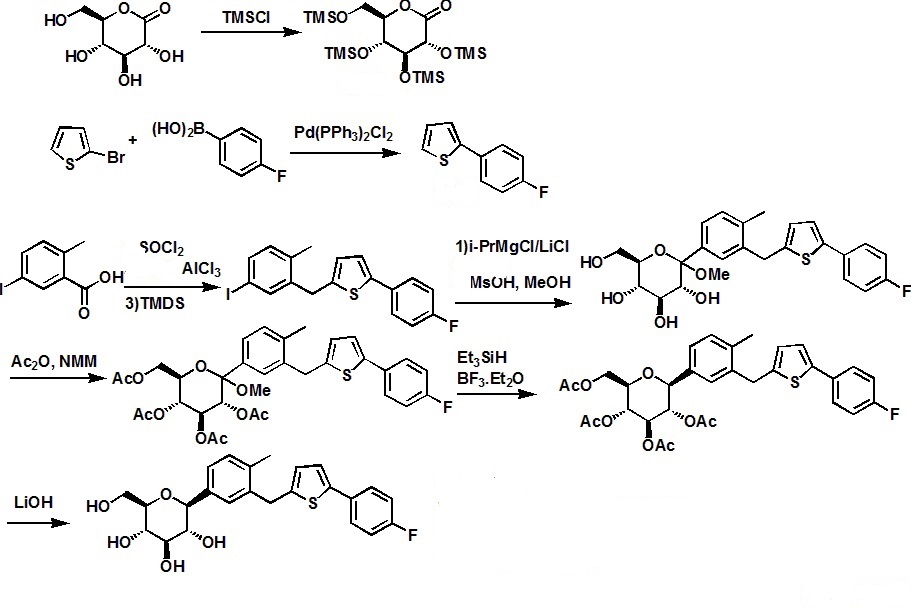
.............
Canagliflozin
is an API that is an inhibitor of SGLT2 and is being developed for the
treatment of type 2 diabetes mellitus.[0011] The IUPAC systematic name
of canagliflozin is (25,,3/?,4i?,55',6 ?)-2-{3-[5-[4-fluoro-
phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol,
and is also known as
(15)-l,5-anhydro-l-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-
methylphenyl]-D-glucitol and l-(
-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-
thienylmethyl]benzene. Canagliflozin is a white to off-white powder with
a molecular formula of C24H25F05S and a molecular weight of 444.52. The structure of canagliflozin is shown as compound B.
 Compound B - Canagliflozin
Compound B - Canagliflozin
[0012] In US 2008/0146515 Al, a crystalline hemihydrate form of canagliflozin (shown as Compound C) is disclosed, having the powder X-ray diffraction (XRPD) pattern comprising the following 2Θ values measured using CuKa radiation: 4.36±0.2, 13.54±0.2, 16.00±0.2, 19.32±0.2, and 20.80±0.2. The XRPD pattern is shown in Figure 24. A process for the preparation of canagliflozin hemihydrate is also disclosed in US 2008/0146515 Al.
 Compound C - hemihydrate form of canagliflozin
Compound C - hemihydrate form of canagliflozin
[0013] In US 2009/0233874 Al, a crystalline form of canagliflozin is disclosed.

[0012] In US 2008/0146515 Al, a crystalline hemihydrate form of canagliflozin (shown as Compound C) is disclosed, having the powder X-ray diffraction (XRPD) pattern comprising the following 2Θ values measured using CuKa radiation: 4.36±0.2, 13.54±0.2, 16.00±0.2, 19.32±0.2, and 20.80±0.2. The XRPD pattern is shown in Figure 24. A process for the preparation of canagliflozin hemihydrate is also disclosed in US 2008/0146515 Al.

[0013] In US 2009/0233874 Al, a crystalline form of canagliflozin is disclosed.
........
WO
2005/012326 pamphlet discloses a class of compounds that are inhibitors
of sodium-dependent glucose transporter (SGLT) and thus of therapeutic
use for treatment of diabetes, obesity, diabetic complications, and the
like. There is described in WO 2005/012326 pamphlet
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
of formula (I):


Example
1 Crystalline
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
hemihydrate1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
was prepared in a similar manner as described in WO 2005/012326.
 (1)
To a solution of
5-bromo-1-[5-(4-fluorophenyl)-2-thienylmethyl]-2-methylbenzene (1, 28.9
g) in tetrahydrofuran (480 ml) and toluene (480 ml) was added
n-butyllithium (1.6M hexane solution, 50.0 ml) dropwise at −67 to −70°
C. under argon atmosphere, and the mixture was stirred for 20 minutes at
the same temperature. Thereto was added a solution of 2 (34.0 g) in
toluene (240 ml) dropwise at the same temperature, and the mixture was
further stirred for 1 hour at the same temperature. Subsequently,
thereto was added a solution of methanesulfonic acid (21.0 g) in
methanol (480 ml) dropwise, and the resulting mixture was allowed to
warm to room temperature and stirred for 17 hours. The mixture was
cooled under ice—water cooling, and thereto was added a saturated
aqueous sodium hydrogen carbonate solution. The mixture was extracted
with ethyl acetate, and the combined organic layer was washed with brine
and dried over magnesium sulfate. The insoluble was filtered off and
the solvent was evaporated under reduced pressure. The residue was
triturated with toluene (100 ml)—hexane (400 ml) to give
1-(1-methoxyglucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]-benzene
(3) (31.6 g). APCI-Mass m/Z 492 (M+NH4).
(1)
To a solution of
5-bromo-1-[5-(4-fluorophenyl)-2-thienylmethyl]-2-methylbenzene (1, 28.9
g) in tetrahydrofuran (480 ml) and toluene (480 ml) was added
n-butyllithium (1.6M hexane solution, 50.0 ml) dropwise at −67 to −70°
C. under argon atmosphere, and the mixture was stirred for 20 minutes at
the same temperature. Thereto was added a solution of 2 (34.0 g) in
toluene (240 ml) dropwise at the same temperature, and the mixture was
further stirred for 1 hour at the same temperature. Subsequently,
thereto was added a solution of methanesulfonic acid (21.0 g) in
methanol (480 ml) dropwise, and the resulting mixture was allowed to
warm to room temperature and stirred for 17 hours. The mixture was
cooled under ice—water cooling, and thereto was added a saturated
aqueous sodium hydrogen carbonate solution. The mixture was extracted
with ethyl acetate, and the combined organic layer was washed with brine
and dried over magnesium sulfate. The insoluble was filtered off and
the solvent was evaporated under reduced pressure. The residue was
triturated with toluene (100 ml)—hexane (400 ml) to give
1-(1-methoxyglucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]-benzene
(3) (31.6 g). APCI-Mass m/Z 492 (M+NH4).
(2) A solution of 3 (63.1 g) and triethylsilane (46.4 g) in dichloromethane (660 ml) was cooled by dry ice-acetone bath under argon atmosphere, and thereto was added dropwise boron trifluoride•ethyl ether complex (50.0 ml), and the mixture was stirred at the same temperature. The mixture was allowed to warm to 0° C. and stirred for 2 hours. At the same temperature, a saturated aqueous sodium hydrogen carbonate solution (800 ml) was added, and the mixture was stirred for 30 minutes. The organic solvent was evaporated under reduced pressure, and the residue was poured into water and extracted with ethyl acetate twice. The organic layer was washed with water twice, dried over magnesium sulfate and treated with activated carbon. The insoluble was filtered off and the solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (300 ml), and thereto were added diethyl ether (600 ml) and H2O (6 ml). The mixture was stirred at room temperature overnight, and the precipitate was collected, washed with ethyl acetate-diethyl ether (1:4) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (33.5 g) as colorless crystals.
mp 98-100° C. APCI-Mass m/Z 462 (M+NH4). 1H-NMR (DMSO-d6) δ 2.26 (3H, s), 3.13-3.28 (4H, m), 3.44 (1H, m), 3.69 (1H, m), 3.96 (1H, d, J=9.3 Hz), 4.10, 4.15 (each 1H, d, J=16.0 Hz), 4.43 (1H, t, J=5.8 Hz), 4.72 (1H, d, J=5.6 Hz), 4.92 (2H, d, J=4.8 Hz), 6.80 (1H, d, J=3.5 Hz), 7.11-7.15 (2H, m), 7.18-7.25 (3H, m), 7.28 (1H, d, J=3.5 Hz), 7.59 (2H, dd, J=8.8, 5.4 Hz).
Anal. Calcd. for C24H25FO5S.0.5H2O: C, 63.56; H, 5.78; F, 4.19; S, 7.07. Found: C, 63.52; H, 5.72; F, 4.08; S, 7.00.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
Example 2An amorphous powder of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene (1.62 g) was dissolved in acetone (15 ml), and thereto were added H2O (30 ml) and a crystalline seed. The mixture was stirred at room temperature for 18 hours, and the precipitate was collected, washed with acetone—H2O (1:4, 30 ml) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (1.52 g) as colorless crystals. mp 97-100° C.

(2) A solution of 3 (63.1 g) and triethylsilane (46.4 g) in dichloromethane (660 ml) was cooled by dry ice-acetone bath under argon atmosphere, and thereto was added dropwise boron trifluoride•ethyl ether complex (50.0 ml), and the mixture was stirred at the same temperature. The mixture was allowed to warm to 0° C. and stirred for 2 hours. At the same temperature, a saturated aqueous sodium hydrogen carbonate solution (800 ml) was added, and the mixture was stirred for 30 minutes. The organic solvent was evaporated under reduced pressure, and the residue was poured into water and extracted with ethyl acetate twice. The organic layer was washed with water twice, dried over magnesium sulfate and treated with activated carbon. The insoluble was filtered off and the solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate (300 ml), and thereto were added diethyl ether (600 ml) and H2O (6 ml). The mixture was stirred at room temperature overnight, and the precipitate was collected, washed with ethyl acetate-diethyl ether (1:4) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (33.5 g) as colorless crystals.
mp 98-100° C. APCI-Mass m/Z 462 (M+NH4). 1H-NMR (DMSO-d6) δ 2.26 (3H, s), 3.13-3.28 (4H, m), 3.44 (1H, m), 3.69 (1H, m), 3.96 (1H, d, J=9.3 Hz), 4.10, 4.15 (each 1H, d, J=16.0 Hz), 4.43 (1H, t, J=5.8 Hz), 4.72 (1H, d, J=5.6 Hz), 4.92 (2H, d, J=4.8 Hz), 6.80 (1H, d, J=3.5 Hz), 7.11-7.15 (2H, m), 7.18-7.25 (3H, m), 7.28 (1H, d, J=3.5 Hz), 7.59 (2H, dd, J=8.8, 5.4 Hz).
Anal. Calcd. for C24H25FO5S.0.5H2O: C, 63.56; H, 5.78; F, 4.19; S, 7.07. Found: C, 63.52; H, 5.72; F, 4.08; S, 7.00.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene
Example 2An amorphous powder of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene (1.62 g) was dissolved in acetone (15 ml), and thereto were added H2O (30 ml) and a crystalline seed. The mixture was stirred at room temperature for 18 hours, and the precipitate was collected, washed with acetone—H2O (1:4, 30 ml) and dried under reduced pressure at room temperature to give 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate (1.52 g) as colorless crystals. mp 97-100° C.
........
there
are a significant number of other β-C-arylglucoside derived drug
candidates, most of which differ only in the aglycone moiety (i.e.,
these compounds comprise a central 1-deoxy-glucose ring moiety that is
arylated at CI). It is this fact that makes them attractive targets for a
novel synthetic platform technology, since a single methodology should
be able to furnish a plurality of products. Among β-C-arylglucosides
that possess known SGLT2 inhibition also currently in clinical
development are canagliflozin, empagliflozin, and ipragliflozin.
 Dapagliflozin Canagliflozin
Dapagliflozin Canagliflozin
 Ipragliflozin .....................Empagliflozin
Ipragliflozin .....................Empagliflozin
[0007] A series of synthetic methods have been reported in the peer-reviewed and patent literature that can be used for the preparation of β-C-arylglucosides. These methods are described below and are referred herein as the gluconolactone method, the metalated glucal method, the glucal epoxide method and the glycosyl leaving group substitution method.
[0008] The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor "redox economy" (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction
 Glucose Gluconoloctone Hemiketal a-anomer β-anomer
Glucose Gluconoloctone Hemiketal a-anomer β-anomer
R = protecting group
[0009] The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
[0010] U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.
 R = protecting group; R' = H or alkyl
R = protecting group; R' = H or alkyl
[0011] The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.
 or
Zn[0012] The glycosyl leaving group substitution method: U.S. Patent
7,847,074, also disclosed a method comprising the substitution of a
leaving group located at CI of a hydroxy-protected glucosyl species,
such as a glycosyl halide, with a metalated aryl compound to prepare
SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar
method to prepare the SGLT2 inhibitor canagliflozin and preferably
diarylzinc complexes are used as nucleophiles along with tetra-
>-pivaloyl protected glucosylbromide.
or
Zn[0012] The glycosyl leaving group substitution method: U.S. Patent
7,847,074, also disclosed a method comprising the substitution of a
leaving group located at CI of a hydroxy-protected glucosyl species,
such as a glycosyl halide, with a metalated aryl compound to prepare
SGLT2 inhibitors. U.S. Pat. Appl. 2011/0087017 disclosed a similar
method to prepare the SGLT2 inhibitor canagliflozin and preferably
diarylzinc complexes are used as nucleophiles along with tetra-
>-pivaloyl protected glucosylbromide.
 Glucose Glucosyl bromide β-anomer
Glucose Glucosyl bromide β-anomer
[0013] Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
[0014] It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
[0015] In view of the above, there remains a need for a shorter, more efficient and
stereoselective, redox economic process for the preparation of β-C-arylglucosides. A new process should be applicable to the industrial manufacture of SGLT2 inhibitors and their prodrugs,
EXAMPLE 22 - Synthesis of 2,4-di-0-feri-butyldiphenylsUyl-l-C-(3-((5-(4- fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (2,4-di-6>-TBDPS- canagliflozin; (IVi"))
 [0227]
2-(5-Bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (1.5 g, 4.15
mmol) and magnesium powder (0.33 g, 13.7 mmol) were placed in a suitable
reactor, followed by THF (9 mL) and 1,2-dibromoethane (95 μί). The
mixture was heated to reflux. After the reaction was initiated, a
solution of 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (2.5
g, 6.92 mmol) in THF (15mL) was added dropwise. The mixture was stirred
for another 2 hours under reflux, and was then cooled to ambient
temperature and titrated to determine the concentration. The thus
prepared 3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl
magnesium bromide (0.29 M in THF, 17 mL, 5.0 mmol) and A1C13
(0.5 M in THF, 4.0 mL, 2.0 mmol) were mixed at ambient temperature to
give a black solution, which was stirred at ambient temperature for 1
hour. To a solution of l
,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64
g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added rc-BuLi
(0.4 mL, 1.0 mmol, 2.5 M solution in Bu20). After stirring
for about 5 min the solution was then added into the above prepared
aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to
rinse the flask. The mixture was concentrated under reduced pressure (50
torr) at 60 °C (external bath temperature) to remove low-boiling point
ethereal solvents, and PhOMe (6 mL) was then added. The remaining
mixture was heated at 150 °C (external bath temperature) for 5 hours at
which time HPLC assay analysis indicated a 68% yield of
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-
(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-
-D-glucopyranoside. After cooling to ambient temperature, the reaction
was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous
earth at ambient temperature, then the mixture was filtered and the
filter cake was washed with THF. The combined filtrates were
concentrated and the crude product was purified by silica gel column
chromatography (eluting with 1 :20 MTBE/rc-heptane) to give the product
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-
methylphenyl)- -D-glucopyranoside (0.51 g, 56%) as a white powder.
[0227]
2-(5-Bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (1.5 g, 4.15
mmol) and magnesium powder (0.33 g, 13.7 mmol) were placed in a suitable
reactor, followed by THF (9 mL) and 1,2-dibromoethane (95 μί). The
mixture was heated to reflux. After the reaction was initiated, a
solution of 2-(5-bromo-2-methylbenzyl)-5-(4-fluorophenyl)thiophene (2.5
g, 6.92 mmol) in THF (15mL) was added dropwise. The mixture was stirred
for another 2 hours under reflux, and was then cooled to ambient
temperature and titrated to determine the concentration. The thus
prepared 3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl
magnesium bromide (0.29 M in THF, 17 mL, 5.0 mmol) and A1C13
(0.5 M in THF, 4.0 mL, 2.0 mmol) were mixed at ambient temperature to
give a black solution, which was stirred at ambient temperature for 1
hour. To a solution of l
,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64
g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added rc-BuLi
(0.4 mL, 1.0 mmol, 2.5 M solution in Bu20). After stirring
for about 5 min the solution was then added into the above prepared
aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to
rinse the flask. The mixture was concentrated under reduced pressure (50
torr) at 60 °C (external bath temperature) to remove low-boiling point
ethereal solvents, and PhOMe (6 mL) was then added. The remaining
mixture was heated at 150 °C (external bath temperature) for 5 hours at
which time HPLC assay analysis indicated a 68% yield of
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-
(4-fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-
-D-glucopyranoside. After cooling to ambient temperature, the reaction
was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous
earth at ambient temperature, then the mixture was filtered and the
filter cake was washed with THF. The combined filtrates were
concentrated and the crude product was purified by silica gel column
chromatography (eluting with 1 :20 MTBE/rc-heptane) to give the product
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)-4-
methylphenyl)- -D-glucopyranoside (0.51 g, 56%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.65 (d, J= 7.2 Hz, 2H), 7.55 (d, J= 7.2 Hz, 2H), 7.48 (dd, J= 7.6, 5.6 Hz, 2H), 7.44-7.20 (m, 16H), 7.11-6.95 (m, 6H), 6.57 (d, J= 3.2 Hz, IH), 4.25 (d, J= 9.6 Hz, IH), 4.06 (s, 2H), 3.90-3.86 (m, IH), 3.81-3.76 (m, IH), 3.61-3.57 (m, IH), 3.54-3.49 (m, 2H), 3.40 (dd, J= 8.8, 8.8 Hz, IH), 2.31 (s, 3H), 1.81 (dd, J= 6.6, 6.6 Hz, IH, OH), 1.19 (d, J= 4.4 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 162.1 (d, J= 246 Hz, C), 143.1 (C), 141.4 (C), 137.9 (C), 136.8 (C), 136.5 (C), 136.4 (CH x2), 136.1 (CH x2), 135.25 (C), 135.20 (CH x2), 135.0 (CH x2), 134.8 (C), 132.8 (C), 132.3 (C), 130.9 (d, J= 3.5 Hz, C), 130.5 (CH), 130.0 (CH), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.6 (CH x4), 127.5 (CH x2), 127.2 (CH x2), 127.1 (d, J= 8.2 Hz, CH x2), 127.06 (CH), 126.0 (CH), 122.7 (CH), 115.7 (d, J= 21.8 Hz, CH x2), 82.7 (CH), 80.5 (CH), 79.4 (CH), 76.3 (CH), 72.9 (CH), 62.8 (CH2), 34.1(CH2), 27.2 (CH3 x3), 26.7 (CH3 x3), 19.6, (C), 19.3 (CH3),19.2 (C); LCMS (ESI) m/z 938 (100, [M+NH4]+), 943 (10, [M+Na]+).
EXAMPLE 23 - Synthesis of canagliflozin (l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)- 4-methylphenyl)- -D-glucopyranoside; (Ii))
 [0228]
A mixture of the
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-
2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (408 mg, 0.44 mmol)
and TBAF (3.5 mL, 3.5 mmol, 1.0 M in THF) was stirred at ambient
temperature for 4 hours. CaC03 (0.73 g), Dowex 50WX8-400 ion
exchange resin (2.2 g) and MeOH (5mL) were added to the product mixture
and the suspension was stirred at ambient temperature for 1 hour and
then the mixture was filtered through a pad of diatomaceous earth. The
filter cake was rinsed with MeOH and the combined filtrates was
evaporated under vacuum and the resulting residue was purified by column
chromatography (eluting with 1 :20 MeOH/DCM) affording canagliflozin (143 mg, 73%).
[0228]
A mixture of the
2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(3-((5-(4-fluorophenyl)thiophen-
2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (408 mg, 0.44 mmol)
and TBAF (3.5 mL, 3.5 mmol, 1.0 M in THF) was stirred at ambient
temperature for 4 hours. CaC03 (0.73 g), Dowex 50WX8-400 ion
exchange resin (2.2 g) and MeOH (5mL) were added to the product mixture
and the suspension was stirred at ambient temperature for 1 hour and
then the mixture was filtered through a pad of diatomaceous earth. The
filter cake was rinsed with MeOH and the combined filtrates was
evaporated under vacuum and the resulting residue was purified by column
chromatography (eluting with 1 :20 MeOH/DCM) affording canagliflozin (143 mg, 73%).
 III II"
III II"
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- "D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).


[0007] A series of synthetic methods have been reported in the peer-reviewed and patent literature that can be used for the preparation of β-C-arylglucosides. These methods are described below and are referred herein as the gluconolactone method, the metalated glucal method, the glucal epoxide method and the glycosyl leaving group substitution method.
[0008] The gluconolactone method: In 1988 and 1989 a general method was reported to prepare C-arylglucosides from tetra-6>-benzyl protected gluconolactone, which is an oxidized derivative of glucose (see J. Org. Chem. 1988, 53, 752-753 and J. Org. Chem. 1989, 54, 610- 612). The method comprises: 1) addition of an aryllithium derivative to the hydroxy-protected gluconolactone to form a hemiketal (a.k.ci., a lactol), and 2) reduction of the resultant hemiketal with triethylsilane in the presence of boron trifluoride etherate. Disadvantages of this classical, but very commonly applied method for β-C-arylglucoside synthesis include:
1) poor "redox economy" (see J. Am. Chem. Soc. 2008, 130, 17938-17954 and Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN- 10: 0120594757); pg 38)— that is, the oxidation state of the carbon atom at CI, with respect to glucose, is oxidized in the gluconolactone and then following the arylation step is reduced to provide the requisite oxidation state of the final product. 2) due to a lack of stereospecificity, the desired β-C-arylglucoside is formed along with the undesired a-C-arylglucoside stereoisomer (this has been partially addressed by the use of hindered trialkylsilane reducing agents (see Tetrahedron: Asymmetry 2003, 14, 3243-3247) or by conversion of the hemiketal to a methyl ketal prior to reduction (see J. Org. Chem. 2007, 72, 9746-9749 and U.S. Patent 7,375,213)).
Oxidation Reduction

R = protecting group
[0009] The metalated glucal method: U.S. Patent 7,847,074 discloses preparation of SGLT2 inhibitors that involves the coupling of a hydroxy-protected glucal that is metalated at CI with an aryl halide in the presence of a transition metal catalyst. Following the coupling step, the requisite formal addition of water to the C-arylglucal double bond to provide the desired C-aryl glucoside is effected using i) hydroboration and oxidation, or ii) epoxidation and reduction, or iii) dihydroxylation and reduction. In each case, the metalated glucal method represents poor redox economy because oxidation and reduction reactions must be conducted to establish the requisite oxidation states of the individual CI and C2 carbon atoms.
[0010] U.S. Pat. Appl. 2005/0233988 discloses the utilization of a Suzuki reaction between a CI -boronic acid or boronic ester substituted hydroxy-protected glucal and an aryl halide in the presence of a palladium catalyst. The resulting 1- C-arylglucal is then formally hydrated to provide the desired 1- C-aryl glucoside skeleton by use of a reduction step followed by an oxidation step. The synthesis of the boronic acid and its subsequent Suzuki reaction, reduction and oxidation, together, comprise a relatively long synthetic approach to C-arylglucosides and exhibits poor redox economy. Moreover, the coupling catalyst comprises palladium which is toxic and therefore should be controlled to very low levels in the drug substance.

[0011] The glucal epoxide method: U.S. Patent 7,847,074 discloses a method that utilizes an organometallic (derived from the requisite aglycone moiety) addition to an electrophilic epoxide located at C1-C2 of a hydroxy-protected glucose ring to furnish intermediates useful for SGLT2 inhibitor synthesis. The epoxide intermediate is prepared by the oxidation of a hydroxy- protected glucal and is not particularly stable. In Tetrahedron 2002, 58, 1997-2009 it was taught that organometallic additions to a tri-6>-benzyl protected glucal-derived epoxide can provide either the a-C-arylglucoside, mixtures of the a- and β-C-arylglucoside or the β-C-arylglucoside by selection of the appropriate counterion of the carbanionic aryl nucleophile (i.e., the
organometallic reagent). For example, carbanionic aryl groups countered with copper (i.e., cuprate reagents) or zinc (i.e., organozinc reagents) ions provide the β-C-arylglucoside, magnesium ions provide the a- and β-C-arylglucosides, and aluminum (i.e., organoaluminum reagents) ions provide the a-C-arylglucoside.


[0013] Methodology for alkynylation of 1,6-anhydroglycosides reported in Helv. Chim. Acta. 1995, 78, 242-264 describes the preparation of l,4-dideoxy-l,4-diethynyl^-D-glucopyranoses (a. La., glucopyranosyl acetylenes), that are useful for preparing but-l,3-diyne-l,4-diyl linked polysaccharides, by the ethynylating opening (alkynylation) of partially protected 4-deoxy-4-C- ethynyl-l,6-anhydroglucopyranoses. The synthesis of β-C-arylglucosides, such as could be useful as precursors for SLGT2 inhibitors, was not disclosed. The ethynylation reaction was reported to proceed with retention of configuration at the anomeric center and was rationalized (see Helv. Chim. Acta 2002, 85, 2235-2257) by the C3-hydroxyl of the 1,6- anhydroglucopyranose being deprotonated to form a C3-0-aluminium species, that coordinated with the C6-oxygen allowing delivery of the ethyne group to the β-face of the an oxycarbenium cation derivative of the glucopyranose. Three molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6-anhydroglucopyranose. The
ethynylaluminium reagent was prepared by the reaction of equimolar (i.e., 1:1) amounts of aluminum chloride and an ethynyllithium reagent that itself was formed by the reaction of an acetylene compound with butyllithium. This retentive ethynylating opening method was also applied (see Helv. Chim. Acta. 1998, 81, 2157-2189) to 2,4-di-<9-triethylsilyl- 1,6- anhydroglucopyranose to provide l-deoxy-l-C-ethynyl- -D-glucopyranose. In this example, 4 molar equivalents of the ethynylaluminium reagent was used per 1 molar equivalent of the 1,6- anhydroglucopyranose. The ethynylaluminium regent was prepared by the reaction of equimolar (i.e., 1: 1) amounts of aluminum chloride and an ethynyl lithium reagent that itself was formed by reaction of an acetylene compound with butyllithium.
[0014] It can be seen from the peer-reviewed and patent literature that the conventional methods that can be used to provide C-arylglucosides possess several disadvantages. These include (1) a lack of stereoselectivity during formation of the desired anomer of the C- arylglucoside, (2) poor redox economy due to oxidation and reduction reaction steps being required to change the oxidation state of CI or of CI and C2 of the carbohydrate moiety, (3) some relatively long synthetic routes, (4) the use of toxic metals such as palladium, and/or (5) atom uneconomic protection of four free hydroxyl groups. With regard to the issue of redox economy, superfluous oxidation and reduction reactions that are inherently required to allow introduction of the aryl group into the carbohydrate moiety of the previously mention synthetic methods and the subsequent synthetic steps to establish the required oxidation state, besides adding synthetic steps to the process, are particular undesirable for manufacturing processes because reductants can be difficult and dangerous to operate on large scales due to their flammability or ability to produce flammable hydrogen gas during the reaction or during workup, and because oxidants are often corrosive and require specialized handling operations (see Anderson, N. G. Practical Process Research & Development, 1st Ed.; Academic Press, 2000 (ISBN-10: 0120594757); pg 38 for discussions on this issue).
[0015] In view of the above, there remains a need for a shorter, more efficient and
stereoselective, redox economic process for the preparation of β-C-arylglucosides. A new process should be applicable to the industrial manufacture of SGLT2 inhibitors and their prodrugs,
EXAMPLE 22 - Synthesis of 2,4-di-0-feri-butyldiphenylsUyl-l-C-(3-((5-(4- fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)- -D-glucopyranoside (2,4-di-6>-TBDPS- canagliflozin; (IVi"))

1H NMR (400 MHz, CDC13) δ 7.65 (d, J= 7.2 Hz, 2H), 7.55 (d, J= 7.2 Hz, 2H), 7.48 (dd, J= 7.6, 5.6 Hz, 2H), 7.44-7.20 (m, 16H), 7.11-6.95 (m, 6H), 6.57 (d, J= 3.2 Hz, IH), 4.25 (d, J= 9.6 Hz, IH), 4.06 (s, 2H), 3.90-3.86 (m, IH), 3.81-3.76 (m, IH), 3.61-3.57 (m, IH), 3.54-3.49 (m, 2H), 3.40 (dd, J= 8.8, 8.8 Hz, IH), 2.31 (s, 3H), 1.81 (dd, J= 6.6, 6.6 Hz, IH, OH), 1.19 (d, J= 4.4 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 162.1 (d, J= 246 Hz, C), 143.1 (C), 141.4 (C), 137.9 (C), 136.8 (C), 136.5 (C), 136.4 (CH x2), 136.1 (CH x2), 135.25 (C), 135.20 (CH x2), 135.0 (CH x2), 134.8 (C), 132.8 (C), 132.3 (C), 130.9 (d, J= 3.5 Hz, C), 130.5 (CH), 130.0 (CH), 129.7 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.6 (CH x4), 127.5 (CH x2), 127.2 (CH x2), 127.1 (d, J= 8.2 Hz, CH x2), 127.06 (CH), 126.0 (CH), 122.7 (CH), 115.7 (d, J= 21.8 Hz, CH x2), 82.7 (CH), 80.5 (CH), 79.4 (CH), 76.3 (CH), 72.9 (CH), 62.8 (CH2), 34.1(CH2), 27.2 (CH3 x3), 26.7 (CH3 x3), 19.6, (C), 19.3 (CH3),19.2 (C); LCMS (ESI) m/z 938 (100, [M+NH4]+), 943 (10, [M+Na]+).
EXAMPLE 23 - Synthesis of canagliflozin (l-C-(3-((5-(4-fluorophenyl)thiophen-2-yl)methyl)- 4-methylphenyl)- -D-glucopyranoside; (Ii))

1H NMR (400 MHz, DMSO-J6) δ 7.63-7.57 (m, 2H), 7.28 (d, J= 3.6 Hz, 1H), 7.23-7.18 (m, 3H), 7.17-7.12 (m, 2H), 6.80 (d, J= 3.6 Hz, 1H), 4.93 (br, 2H, OH), 4.73 (br, 1H, OH), 4.44 (br,IH, OH), 4.16 (d, J= 16 Hz, 1H), 4.10 (d, J= 16 Hz, 1H), 3.97 (d, J= 9.2 Hz, 1H), 3.71 (d, J=I I.6 Hz, 1H), 3.47-3.43 (m, 1H), 3.30-3.15 (m, 4H), 2.27 (s, 3H);
13C NMR (100 MHz, DMSO- d6) δ 161.8 (d, J= 243 Hz, C), 144.1 (C), 140.7 (C), 138.7 (C), 137.8 (C), 135.4 (C), 131.0 (d, J= 3.1 Hz, C), 130.1 (CH), 129.5 (CH), 127.4 (d, J= 8.1 Hz, CH x2), 126.8 (CH), 126.7 (CH), 123.9 (CH), 116.4 (d, J= 21.6 Hz, CH x2), 81.8 (CH), 81.7 (CH), 79.0 (CH), 75.2 (CH), 70.9 (CH), 61.9 (CH2), 33.9 (CH2), 19.3 (CH3);
LCMS (ESI) m/z 462 (100, [M+NH4]+), 467 (3, [M+Na]+).
Example 1 - Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II")
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- "D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
........
FIG. 1:
X-ray powder diffraction pattern of the crystalline of hemihydrate of the compound of formula (I).
X-ray powder diffraction pattern of the crystalline of hemihydrate of the compound of formula (I).

FIG. 2:
Infra-red spectrum of the crystalline of hemihydrate of the compound of formula (I).http://www.google.com/patents/US7943582
Infra-red spectrum of the crystalline of hemihydrate of the compound of formula (I).http://www.google.com/patents/US7943582

.............
FIGS. 3 and 4 provide the XRPD pattern and IR spectrum, respectively, of amorphous canagliflozin.

..................



 | |
| Systematic (IUPAC) name | |
|---|---|
(2S,3R,4R,5S,6R)-2-{3-[5-[4-Fluoro-phenyl)-thiophen-2-ylmethyl]-4-methyl-phenyl}-6-hydroxymethyl-tetrahydro-pyran-3,4,5-triol
| |
| Clinical data | |
| Trade names | Invokana |
| AHFS/Drugs.com | entry |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 65% |
| Protein binding | 99% |
| Metabolism | Hepatic glucuronidation |
| Biological half-life | 11.8 (10–13) hours |
| Excretion | Fecal and 33% renal |
| Identifiers | |
| CAS Registry Number | 842133-18-0 |
| ATC code | A10BX11 |
| PubChem | CID: 24812758 |
| IUPHAR/BPS | 4582 |
| DrugBank | DB08907 |
| ChemSpider | 26333259 |
| UNII | 6S49DGR869 |
| ChEBI | CHEBI:73274 |
| ChEMBL | CHEMBL2103841 |
| Synonyms | JNJ-28431754; TA-7284; (1S)-1,5-anhydro-1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-D-glucitol |
| Chemical data | |
| Formula | C24H25FO5S |
| Molecular mass | 444.52 g/mol |
1H NMR PREDICT



13C NMR PREDICT


COSY PREDICT

update........

CAS 1672658-93-3
C24 H25 F O6 S, 460.52
D-Glucopyranose, 1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-

CAS 1809403-04-0
C24 H25 F O6 S, 460.52
D-Glucose, 1-C-[3-[[5-(4-fluorophenyl)-2-thienyl]methyl]-4-methylphenyl]-
WO2017/93949

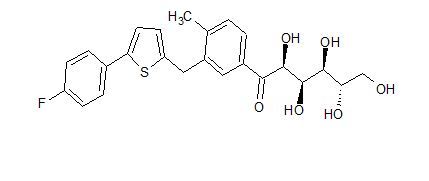
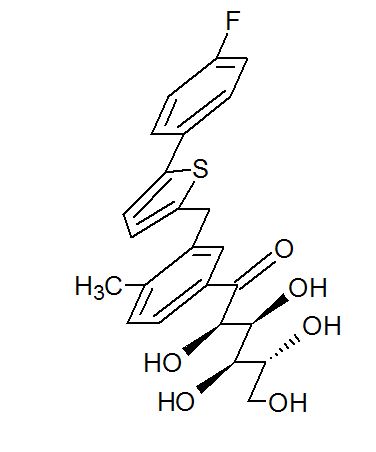
(2R,3S,4R,5R)-1-(3-((5-(4-Fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2,3,4,5,6-pentahydroxyhexan-1-one 12
From the FT-IR spectra of 12 contain a signal at 1674 cm–1, this signal is strongly indicative of a carbonyl ketone being present in 12
In 13C NMR and HMBC correlations spectra, the chemical shift at 199.75 ppm was observed. Analysis of the NMR data confirmed that the structure of 12 is a ring-opened keto form
Synthesis of (2R,3S,4R,5R)-1-(3-((5-(4-Fluorophenyl)thiophen-2-yl)methyl)-4-methylphenyl)-2,3,4,5,6-pentahydroxyhexan-1-one 12
title compound 12 (84.23% yield) and having 99.4% purity by HPLC;
DSC: 160.84–166.44 °C;
Mass: m/z 459 (M+–H);
IR (KBr, cm–1): 3313, 2982, 1674.7, 1601, 1507.5, 1232.7;
1H NMR (600 MHz, DMSO-d6) δ 7.87 (s, 1H), 7.80 (dd, J = 1.8 Hz, 1H), 7.61–7.58 (m, 2H), 7.33 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 3.6 Hz, 1H), 7.21–7.18 (m, 2H), 6.84 (d, J = 3.6 Hz, 1H), 5.17 (dd, J = 3.6, 3.0 Hz, 1H), 5.02 (d, J = 6.6 Hz, 1H), 4.57 (d, J = 4.8 Hz, 1H), 4.43–4.39 (m, 3H), 4.22 (s, 2H), 4.02–4.01 (m, 1H), 3.53–3.51 (m, 3H), 3.38–3.37 (m, 1H), 2.35 (s, 3H);
13C NMR (101 MHz, DMSO-d6) δ 199.7, 162.6, 160.2, 142.8, 142.1, 140.5, 138.8, 133.3, 130.5, 130.4, 130.4, 129.3, 127.2, 127.0, 127.0, 126.7, 123.5, 116.0, 115.8, 75.2, 72.3, 71.8, 71.3, 63.2, 33.2, 19.2.
HRMS (ESI): calcd m/zfor [C24H25FO6S + Na]+ = 483.1248, found m/z 483.1244.













Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.7b00281
////////////
References
- New J&J diabetes drug effective in mid-stage study, Jun 26, 2010
- Edward C. Chao (2011). "Canagliflozin". Drugs of the Future 36 (5): 351–357. doi:10.1358/dof.2011.36.5.1590789.
- http://www.investor.jnj.com/releasedetail.cfm?releaseid=710584
- "Summary Review" (PDF). FDA.gov. March 29, 2013. Retrieved 2014-07-09.
- "HIGHLIGHTS OF PRESCRIBING INFORMATION" (PDF). FDA.gov. 2013. Retrieved 2014-07-09.
- Haberfeld, H (ed.). Austria-Codex (in German) (2013/14, supplement 01/14 ed.). Vienna: Österreichischer Apothekerverlag.
- FDA (2015-05-15). "SGLT2 inhibitors: Drug Safety Communication - FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood". Retrieved 19 May 2015.
- http://www.medscape.com/viewarticle/777503
- Balis, Dainius A; Tong, Cindy; Meininger, Gary (July 2014). "Effect of canagliflozin, a sodium–glucose cotransporter 2 inhibitor, on measurement of serum 1,5-anhydroglucitol". J Diabetes 6 (4): 378–380. doi:10.1111/1753-0407.12116.
- Prous Science: Molecule of the Month November 2007
- A. Klement (20 January 2014). "Tubuläre Senkung des Blutzuckers bei Diabetes mellitus: Invokana". Österreichische Apothekerzeitung (in German) (2/2014): 20f.
- "EMEA-001030-PIP01-10". EMA European Medicines Agency. Retrieved May 6, 2013.
- "U.S. FDA approves Johnson & Johnson diabetes drug, canagliflozin". Reuters. Mar 29, 2013.
U.S. health regulators have approved a new diabetes drug from Johnson & Johnson, making it the first in its class to be approved in the United States.
| WO2005012326A1 | Jul 30, 2004 | Feb 10, 2005 | Tanabe Seiyaku Co | Novel compounds having inhibitory activity against sodium-dependant transporter |
| WO2013064909A2 * | Oct 30, 2012 | May 10, 2013 | Scinopharm Taiwan, Ltd. | Crystalline and non-crystalline forms of sglt2 inhibitors |
| CN103655539A * | Dec 13, 2013 | Mar 26, 2014 | 重庆医药工业研究院有限责任公司 | Oral solid preparation of canagliflozin and preparation method thereof |
| US7943582 | Dec 3, 2007 | May 17, 2011 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyransoyl)-4-methyl-3-[5-(4-fluorophenyl)-2- thienylmethyl]benzene hemihydrate |
| US7943788 | Jan 31, 2005 | May 17, 2011 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US8513202 | May 9, 2011 | Aug 20, 2013 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate |
| US20130237487 | Oct 30, 2012 | Sep 12, 2013 | Scinopharm Taiwan, Ltd. | Crystalline and non-crystalline forms of sglt2 inhibitors |
| WO2008002824A1 * | Jun 21, 2007 | Jan 3, 2008 | Squibb Bristol Myers Co | Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetes |
| US6774112 * | Apr 8, 2002 | Aug 10, 2004 | Bristol-Myers Squibb Company | Amino acid complexes of C-aryl glucosides for treatment of diabetes and method |
| US20090143316 * | Apr 4, 2007 | Jun 4, 2009 | Astellas Pharma Inc. | Cocrystal of c-glycoside derivative and l-proline |
| US20110087017 * | Oct 14, 2010 | Apr 14, 2011 | Vittorio Farina | Process for the preparation of compounds useful as inhibitors of sglt2 |
| US20110098240 * | Aug 15, 2008 | Apr 28, 2011 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition comprising a sglt2 inhibitor in combination with a dpp-iv inhibitor |
| Reference | ||
|---|---|---|
| 1 | * | OGURA H. ET AL.: '5-FLUOROURACIL NUCLEOSIDES. SYNTHESIS OF A STEREO-CONTROLLED NUCLEOSIDE SYNTHESIS FROM ANHYDRO SUGARS' NUCLEIC ACID CHEM. vol. 4, 1991, pages 109 - 112, XP000607288 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2014195966A2 * | May 30, 2014 | Dec 11, 2014 | Cadila Healthcare Limited | Amorphous form of canagliflozin and process for preparing thereof |
| US9006188 | May 23, 2014 | Apr 14, 2015 | Mapi Pharma Ltd. | Co-crystals of dapagliflozin |
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one
in a million spine stroke called acute transverse mylitis, it made me
90% paralysed and bound to a wheel chair, Now I keep him as my source of
inspiration and helping millions, thanks to millions of my readers who
keep me going and help me to keep my son happy

///////////
TAKE A TOUR
Sveti Stefan, MONTENEGRO

Sveti Stefan - Wikipedia, the free encyclopedia
https://en.wikipedia.org/wiki/Sveti_Stefan


////////////