
RIVAROXABAN

5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide (Rivaroxaban) (1):1
rivaroxaban 1 (689 mg) in 88% yield, Rf = 0.30 (ethyl acetate), as a white solid,
m.p. 229.3–230.7 °C(lit.1, 230 °C).
[α]D20 = −37° (c = 0.5, DMSO) [lit.1, [α]D21 = –38°(c = 0.2985, DMSO)].
IR (KBr) (νmax /cm−1): 3343, 1724 (C=O), 1649(C=O), 1523, 1430, 808, 756.
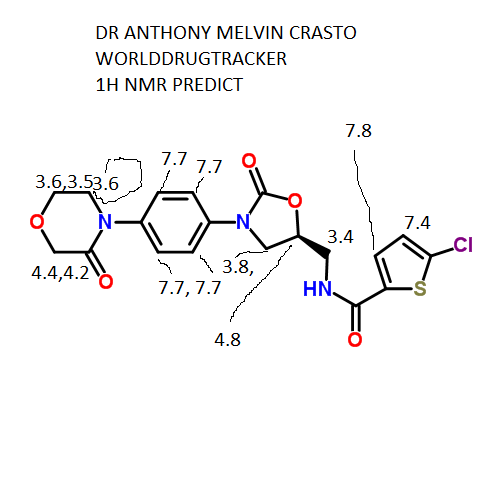
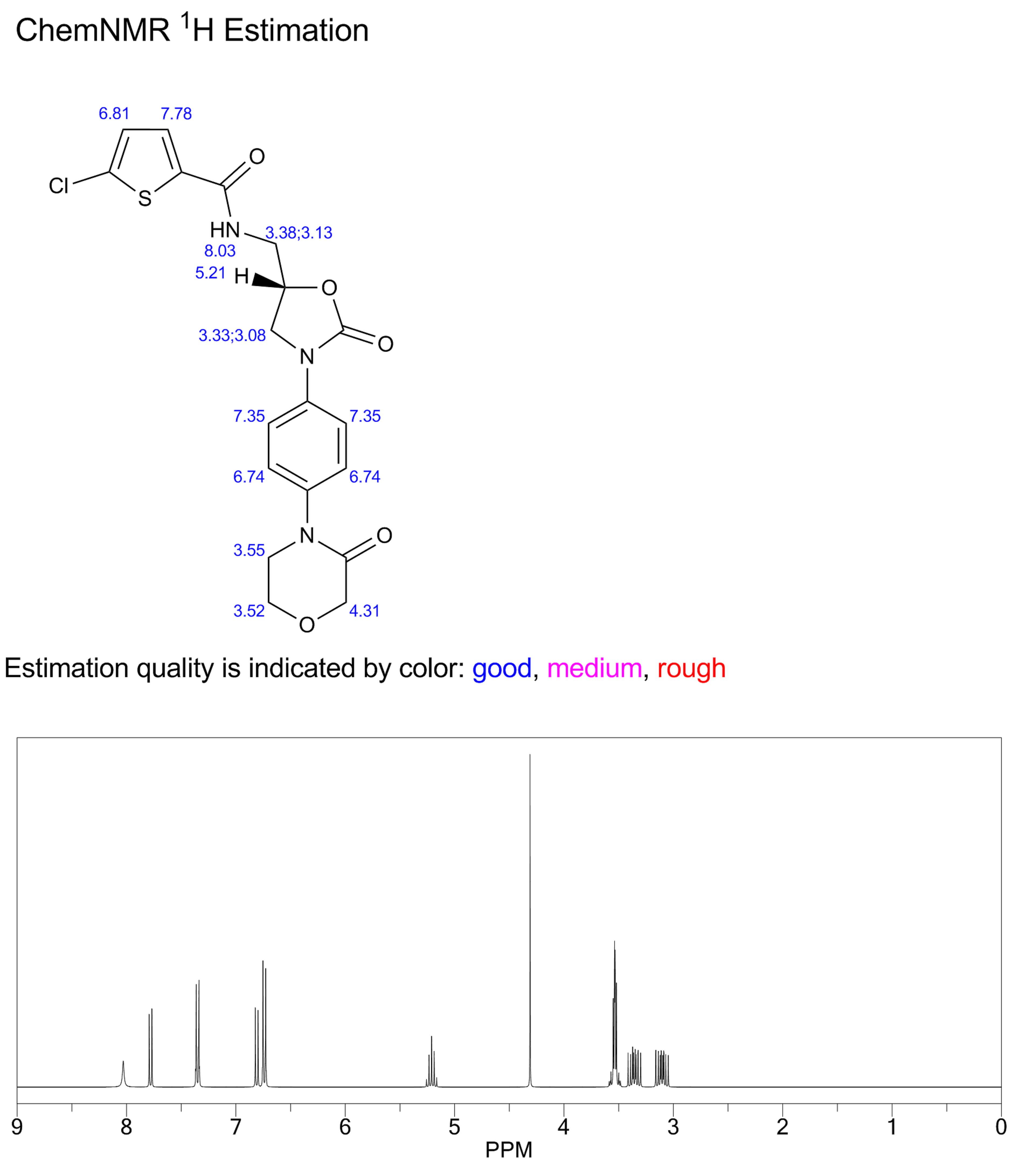
δH 3.60–3.62 (m, 2H), 3.71–3.73 (m,2H), 3.84–3.87 (dd, J = 6.5, 9.5 Hz, 1H), 3.96–3.98 (m, 2H), 4.20 (s,2H), 4.18–4.21 (m, 1H), 4.83–4.86 (m, 1H), 7.20 (d, J = 4.0 Hz, 1H),7.41 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 9.0 Hz, 2H), 7.69 (d, J = 4.0 Hz,1H), 8.99 (t, J = 5.5 Hz, 1H).
δC 42.19, 47.43, 49.00, 63.46, 67.71,71.30, 118.35, 125.92, 128.11, 128.43, 133.24, 136.48, 137.08,138.43, 154.08, 160.79, 165.95.
JOURNAL OF CHEMICAL RESEARCH v 35, issue 7, pg 400-4-1, 2011
An approach to the anticoagulant agent rivaroxaban via an isocyanate-oxirane cycloaddition promoted by MgI2.etherate
Chao Lia, Yingshuai Liua, Yongjun Zhangb and Xingxian Zhanga*
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
A convergent and efficient synthesis of anticoagulant rivaroxaban was developed using the cycloaddition of commercially
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
* Correspondent. E-mail: mhmosslemin@yahoo.com
REF 1=S. Roehrig, A. Straub, J. Pohlmann, T. Lampe, J. Pernerstorfer, K.Schlemmer, P. Reinemer and E. Perzborn, J. Med. Chem., 2005, 48, 5900.
.........................
 RIVAROXABAN
RIVAROXABAN
IH NMR PREDICT

RIVAROXABAN

.........................
RIVAROXABANIH NMR PREDICT
RIVAROXABAN
..............................
13 C NMR PREDICT


.
13 C NMR PREDICT
.
EXAMPLE 28 (preparation of rivaroxaban)

10 g of the salt prepared according to Example 18 were suspended in 75 ml of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of triethylamine was added and the mixture was heated at 60°C. This was followed by addition of 15.7 ml of a solution of 5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the reaction mixture was stirred and heated at 55°C for 15 minutes, then slowly cooled below 30°C, 75 ml were added and the turbid solution was filtered. The clear filtrate was stirred at 50°C, which was followed by addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour under slow cooling. The separated product was filtered off, washed with water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g (yield 81%) of rivaroxaban in the form of an off-white powder with the melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the ( )-isomer below 0.03%. 1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2x1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)

The optical isomer of rivaroxaban with the (R)- configuration was obtained by a process analogous to Example 28 starting from the salt prepared according to Example 19. The yield was 76%, HPLC 99.90%, content of the (5)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.
EXAMPLE 30 (preparation of rivaroxaban)

10.5 g of the amine prepared according to Example 26 were suspended in 200 ml of dichloromethane and then 5.4 ml of triethylamine dissolved in 50 ml of dichloromethane were added. This was followed by addition of 14.4 ml of a solution of 5-chlorothiophene-2- carboxylic acid chloride in toluene (2.46 M) and 25 ml of dichloromethane. The reaction mixture was stirred and heated at boiling for 1.5 hours and then slowly cooled below 30°C. The separated product was filtered off, washed with dichloromethane (15 ml) and ethanol (2 x 15 ml). The crude product was crystallized from a mixture of acetic acid (20 ml) and ethanol (200 ml). 10.5 g (yield 67%) of rivaroxaban was obtained in the form of an off-white powder with the melt, point of 228-230°C, HPLC 99.97%, content of the (i?)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.

20 g of 2-({(5S -2-oxo-3-[4-(3-oxomo holin-4-yl) henyl]-l ,3-oxazolidin-5-yl}methyl)-lH- isoindole-l,3(2H)-dione was suspended in 450 ml of ethanol, which was followed by addition of 6 ml of hydrazinehydrate in 50 ml of ethanol and the mixture was stirred and refluxed for 3 hours. Then, the suspension was cooled down to 65°C and filtered and the cake was washed with 2x50 ml of nearly boiling ethanol. After drying 7.2 g of an off-white powder were obtained, which slowly melted at a temperature over 260°C (this fraction contained 99.9% of 2,3-dihydrophtalazine-l,4-dione according to HPLC). After cooling of the hot filtrate another solid fraction separated. After its isolation by filtration and drying, 12.5 g of white lumpy powder with the melt, point of 141-144°C was obtained (this fraction contained 86.7% of the desired product, 12.7% of 2,3-dihydrophtalazine-l,4-dione, and the rest to 100 % were other unidentifiable impurities according to HPLC). The yield of the isolated amine calculated to the pure substance was 73%.

(S)-isomer 99,79 % (R)-isomer 0,21 % (S)-isomer 99,81 % / (R)-isomer 0,19 %
Crude rivaroxaban with the chemical purity of 99.5%, containing 0.21% of the (i?)-enantiomer and 0.50% of unidentified impurities, was crystallized from a mixture of acetic acid and ethanol.
Crystallization 1: 22 g of crude rivaroxaban were dissolved in 180 ml of acetic acid at boiling and the obtained solution was still hot filtered. The filtrate was brought to boil again and 400 ml of ethanol was gradually added to the boiling solution. The mixture was stirred under slow cooling for ca. 1 hour (resulting temperature of the suspension ca. 28°C). Subsequently, filtration was performed, the cake was washed with 2x30 ml of ethanol and vacuum-dried. 20.3 g of the product was obtained, melt, point 227.5-228.5°C. The yield of the crystallization was 92%, HPLC 99.8%, content of the ( ?)-enantiomer 0.21%, contents of unidentified impurities 0.15%.
Crystallization 2: 20 g of once crystallized rivaroxaban were dissolved in 150 ml of acetic acid at boiling and the obtained solution was still hot filtered (the filter was washed with 20 ml of boiling acetic acid to the filtrate). The filtrate was brought to boil again and 340 ml of ethanol was gradually added to the boiling solution. The mixture was stirred under slow cooling for ca. 1.5 hours (resulting temperature of the suspension ca. 26°C). Subsequently, filtration was performed, the cake was washed with 2x50 ml of ethanol and vacuum-dried. 19.1 g of the product was obtained, melt, point 230-231 °C. Crystallization yield 96%, HPLC 99.96%, content of the (fl)-enantiomer 0.19%, the contents of unidentified impurities was 0.04%.
After two crystallizations the final chemical purity of rivaroxaban achieved was 99.96%, the contents of unidentified impurities was reduced from 0.50% to 0.04%. After two crystallizations the achieved content of the (/?)-isomer was 0.19%, which is an excessive value (limit 0.15%) and is comparable to the initial level of 0.21%. The yield of rivaroxaban after the two crystallizations was 88% (calculated to the starting crude rivaroxaban).
.............................................................................................
 COSY NMR
COSY NMR.
13 C NMR.....http://www.nmrdb.org/13c/index.shtml?v=v2.14.1
CLICK TO PREDICT..ALLOW SOME TIME TO LOAD ON NMRDB SITE.....CHECK JAVA AND FLASH SETTINGS
ABOVE PICTURES ARE THE ONES YOU WILL GET

SYNTHESIS
SYN 1

BAYER HEALTHCARE AG Patent: WO2004/60887 A1, 2004 ; Location in patent: Page/Page column 8; 10-11 ;
SYN 2


MEDICHEM S.A.; MANGION, Bernardino; DURAN LOPEZ, Ernesto Patent: WO2012/35057 A2, 2012 ; Location in patent: Page/Page column 34 ;
SYN 3

EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 49 ;
SYN4
EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 68 ;
SYN 5
INTERQUIM, S.A.; Berzosa Rodríguez, Xavier; Marquillas Olondriz, Francisco; Llebaria Soldevilla, Amadeo; Serra Comas, Carme Patent: US2014/128601 A1, 2014 ; Location in patent: Paragraph 0068 ;
SYN 6
MEGAFINE PHARMA (P) LTD; MATHAD Vijayavitthal Thippannachar; PATIL NILESH SUDHIR, Nilesh; NIPHADE NAVNATH CHINTAMAN, Navnath; MALI ANIL CHATURLAL, Anil; BODAKE MAHENDRA BHAGIRATH, Mahendra; IPPAR SHARAD SUBHASH, Sharad; TALLA RAJESH, Rajesh Patent: WO2013/121436 A2, 2013 ; Location in patent: Page/Page column 31 ;
............................
PATENT
The drug compound having the adopted name “Rivaroxaban” has chemical name, 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5- yljmethyl)-2-thiophenecarboxamide; and has the structural formula I,

Formula I
The commercial pharmaceutical product XARELTO® tablets, contains rivaroxaban as active ingredient. Rivaroxaban is a factor Xa inhibitor useful as oral anticoagulant. Rivaroxaban can be used for the prevention and treatment of various thromboembolic diseases, in particular of deep vein thrombosis (DVT), pulmonary embolism (PE), myocardial infract, angina pectoris and restenoses after angioplasty or aortocoronary bypass, cerebral stroke, transitory ischemic attacks, and peripheral arterial occlusive diseases.
U.S. Patent No. 7, 157,456 describes Rivaroxaban and process for the preparation thereof. The process of US ‘456 for rivaroxaban involves reaction of 2-[(2S)-2-oxiranylmethyl]-lH-isoindole-l,3(2H)-dione with 4-(4-aminophenyl)-3-morpholinone to provide 2-((2R)-2-hydroxy-3- { [4-(3-oxo-4-morpholiny)phenyl]amino Jpropyl)- lH-isoindole- 1 ,3(2H)-dione, which on cyclization using Ν,Ν-carbonyl diimidazole to afford 2-({5S)-2-Oxo-3-[4-(3-oxo-4-morpholiny)phenyl]-l,3-oxazolidin-5-yl}methyl)-lH-isoindole-l,3(2H)-dione, which on reacted with methylamine followed by reaction with 5-chlorothiophene-2-carbonyl chloride to provide Rivaroxaban.
Various processes for the preparation of rivaroxaban, its intermediates, and related compounds are disclosed in U.S. Patent Nos. 7,585,860; 7,351,823, 7,816,355, and 8,101,609; patent application Nos. WO 2011/012321, WO 2012/156983, WO 2012/153155, WO 2013/053739, WO 2013/098833, WO 2013/156936, WO 2013/152168, WO 2013/120464, WO 2013/164833, US 2012/0283434 and US 2013/184457; and J. Med. Chem. 2005, 48, 5900-5908.
New patent WO-2015104605
Process for preparing rivaroxaban – comprising the reaction of a thioester compound and its salts with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one.
Wockhardt Ltd
The synthesis of (II) via intermediate (I) is described (example 7, page 15)
4-{4-[(5S)-5-(Aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one (formula III) is (I) and rivaroxaban is (II) (claim 1, page 16).
The present invention relates to a process for the preparation of Rivaroxaban and its novel intermediates, or pharmaceutically acceptable salts thereof. The present invention provides novel intermediates, which may be useful for the preparation of Rivaroxaban or its pharmaceutically acceptable salts thereof. The process of preparation by using novel intermediate is very simple cost effective and may be employed at commercial scale. The product obtained by using novel intermediate yield the Rivaroxaban of purity 99% or more, when measured by HPLC. The present invention especially relates to a process for the preparation of Rivaroxaban from thioester of formula II, or a pharmaceutically acceptable salt thereof, wherein R is leaving group.
process includes the step of , reacting thioester of formula IIA or pharmaceutically acceptable salt thereof
Formula IIA

with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one of formula III,
Formula III

Formula I
EXAMPLE 7: One pot process for Rivaroxaban
The triphenylphosphine (11.5g) and mercaptobenzothiazole disulphide (15.31g) were taken in methylene chloride and reaction mixture was stirred at 28°C -30°C for 1 hr. The 5-chlorothiophene-2-carboxylic acid (7.2g) and triethylamine (3.8 g) were added to the above reaction mixture. The reaction mixture is stirred at 0°C -25 °C for 1 hr. after 1 hr 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one (lOg) and triethylamine (3.8g) were added. The resulting reaction mixture further stirred for 2 hrs. After completion of the reaction, water was added and stirred for 10 min. aqueous layer was separated and washed with methylene chloride. The organic layer was acidified to pH 6-7 with 2N hydrochloric acid and finally the organic layer was concentrated to get desired product. The product was purified and dried to yield Rivaroxaban.
Yield: 10.0 gm
Purity: 99.3 %
EXAMPLE 8: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent ethyl acetate and base potassium hydroxide were used to get the rivaroxaban.
EXAMPLE 9: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent acetonitile and base potassium carbonate were used, methylene chloride was added in the reaction mixture to extract the Rivaroxaban.
.........................
http://www.google.com/patents/WO2013120465A1?cl=en
Rivaroxaban, chemically (S)-5-chloro-N-({2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-l,3- oxazolidin-5-yl}methyl)thiophene-2-carboxamide, described by formula (1), was developed by the company Bayer Healthcare (WO 01/47919, 2001). Rivaroxaban is applied in the clinical practice as the active ingredient of an orally available anticoagulant that is commercially marketed as Xarelto and is used in the prevention and treatment of arterial or venous thromboembolic disorders. In its effect, rivaroxaban is characterized by direct selective inhibition of the FXa coagulation enzyme (Drugs of the Future 2006, 31(6): 484-493).
H2

For the preparation of rivaroxaban several key structures, referred to as building blocks, can be used as advanced intermediates. Virtually all the so far described syntheses are using two such building blocks. The first one are derivatives of 4-(4-aminophenyl)morpholin-3-one, where it may be the case of an unsubstituted amine (2, G means hydrogen), or a derivative alkylated on nitrogen, or a carbamate derived from this compound (2, G means an alkyl or COOalkyl group). The other general and commonly used building block for the rivaroxaban molecule are derivatives of 5-chlorothiophene-2-carboxylic acid (3, X means -OH), or its functional derivatives such as the chloride and amide (3, X means -CI or -NH2). Various synthetic approaches used for synthesis of rivaroxaban differ from each other mainly as regards the chiral building block, which is the source for the construction of the central heterocycle, i.e., 2-oxo-l,3-oxazolidine, wherein the chirality centre is also located. For pharmaceutical purposes one optical isomer derived from rivaroxaban is only used, in particular the target molecule with the absolute configuration (5)-. The selection of a suitable chiral building block must be subjected to this fact.

(4) (5) (6) (7) (8)
Chiral building blocks that have been successfully used for synthesis of rivaroxaban include (5)-glycidyl phthalimide (4), (S)-3-aminopropane-l,2-diol (5), ( ?)-epichlorohydrin (6) and (i?)-glycidyl butyrate (7). (S)-glycidol (8) was used as a starting material for the preparation of (5)-glycidyl phthalimide (4) (Tetrahedron: Asymmetry, Vol. 7, No. 6, pp. 1641-1648, 1996).
The known methods of chemical synthesis of rivaroxaban (1) are described in Schemes 1 to 7. The first one is the process according to Scheme 1 (WO 01/47919 Bayer, US 7 157 456 B2, J.MedChem. (2005), 48(19), 5900-5908), which starts from 4-(4-aminophenyl)morpholin-3- one and (5)-glycidyl phthalimide (4). The second synthetic process follows Scheme 2 (WO 2004/060887, Bayer) and starts from 5-chlorothiophene-2-carboxylic acid (3, X means -OH) and (5)-3-aminopropane-l,2-diol (5). 4-(4-aminophenyl)morpholin-3-one only engages in the synthesis in the penultimate stage in case of the process according to Scheme 2.

Scheme 1

The third synthetic process, which proceeds according to Scheme 3, was mainly used for preparation of deuterated analogs of rivaroxaban (WO 2009/023233 Al, Concert Pharm.). It also represents the first synthetic process in which (i?)-epichlorohydrin (6) was used as the chiral building block. The other key starting material for the third process was 4-(4- aminophenyl)morpholin-3-one. The fourth synthetic process, which proceeds according to Scheme 4 (WO 2010/124835 Al, Apotex), again uses (i?)-epichlorohydrin as the chiral building block, which reacts with the alkyl carbamate derived from 4-(4- aminophenyl)morpholin-3-one in the key stage. The fifth synthetic process, which proceeds according to Scheme 5 (US 20110034465 Al), also uses (i?)-epichlorohydrin as the chiral building block, which directly reacts with 4-(4-aminophenyl)morpholin-3-one in the key stage, which is the same reaction as in the third process. The differences between the third and fifth processes consist in the preparation method of the 2-oxo-l,3-oxazolidine cycle and in the carbonylation agent used. While the third process uses Ι,Γ-carbonyldiimidazol (CDI) as the carbonylation agent, the fifth process uses more available and cheaper alkyl chloroformates.

Scheme 3

Scheme 4

Scheme 5
The sixths synthetic process, which proceeds according to Scheme 6 (WO 2011/080341 Al), uses (7?)-glycidyl butyrate (7) as the chiral building block, which in the key stage reacts with the alkyl carbamate derived from 4-(4-aminophenyl)morpholin-3-one. The last, seventh synthetic process leading to rivaroxaban proceeds according to Scheme 7 (WO 201 1/098501 Al) and, like process 2, uses (S)-3-aminopropane-l,2-diol (5) as the chiral building block. The differences between the second and seventh processes consist in the preparation process of the 2-oxo-l,3-oxazolidine cycle and the carbonylation agent used. While the second process uses Ι,Γ-carbonyldiimidazol (CDI) as the carbonylation agent, the fifth process uses the cheaper, but very toxic phosgene.

Scheme 6

Scheme 7 The processes used for the synthesis of rivaroxaban differ from each other especially in the chiral building block (compounds 4 to 7) and in the carbonylation agents (CDI, alkyl chloroformates, phosgene) used. Another difference can be found in the method of performing deprotection reactions, i.e. such reactions that lead to elimination of the protecting groups, initially bound to the nitrogen atom of the advanced intermediates and which had the initial purpose of protecting these intermediates from undesired chemical transformations. No deprotection reactions were necessary in the case of the processes according to Schemes 2, 4 and 7, as the protecting groups bound to the nitrogen atom eventually became part of the final product. In the case of process 6 it was necessary to deprotect the fert-butyl group bound to the nitrogen. The reaction used was an acid catalyzed reaction of the tert-butyl group, releasing isobutylene according to Scheme 8. In normal conditions isobutylene is a gas and thus can be very easily separated from the final product.

isobutylen

EXAMPLE 28 (preparation of rivaroxaban)
10 g of the salt prepared according to Example 18 were suspended in 75 ml of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of triethylamine was added and the mixture was heated at 60°C. This was followed by addition of 15.7 ml of a solution of 5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the reaction mixture was stirred and heated at 55°C for 15 minutes, then slowly cooled below 30°C, 75 ml were added and the turbid solution was filtered. The clear filtrate was stirred at 50°C, which was followed by addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour under slow cooling. The separated product was filtered off, washed with water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g (yield 81%) of rivaroxaban in the form of an off-white powder with the melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the ( )-isomer below 0.03%. 1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2x1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)
The optical isomer of rivaroxaban with the (R)- configuration was obtained by a process analogous to Example 28 starting from the salt prepared according to Example 19. The yield was 76%, HPLC 99.90%, content of the (5)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.
EXAMPLE 30 (preparation of rivaroxaban)
10.5 g of the amine prepared according to Example 26 were suspended in 200 ml of dichloromethane and then 5.4 ml of triethylamine dissolved in 50 ml of dichloromethane were added. This was followed by addition of 14.4 ml of a solution of 5-chlorothiophene-2- carboxylic acid chloride in toluene (2.46 M) and 25 ml of dichloromethane. The reaction mixture was stirred and heated at boiling for 1.5 hours and then slowly cooled below 30°C. The separated product was filtered off, washed with dichloromethane (15 ml) and ethanol (2 x 15 ml). The crude product was crystallized from a mixture of acetic acid (20 ml) and ethanol (200 ml). 10.5 g (yield 67%) of rivaroxaban was obtained in the form of an off-white powder with the melt, point of 228-230°C, HPLC 99.97%, content of the (i?)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.
20 g of 2-({(5S -2-oxo-3-[4-(3-oxomo holin-4-yl) henyl]-l ,3-oxazolidin-5-yl}methyl)-lH- isoindole-l,3(2H)-dione was suspended in 450 ml of ethanol, which was followed by addition of 6 ml of hydrazinehydrate in 50 ml of ethanol and the mixture was stirred and refluxed for 3 hours. Then, the suspension was cooled down to 65°C and filtered and the cake was washed with 2x50 ml of nearly boiling ethanol. After drying 7.2 g of an off-white powder were obtained, which slowly melted at a temperature over 260°C (this fraction contained 99.9% of 2,3-dihydrophtalazine-l,4-dione according to HPLC). After cooling of the hot filtrate another solid fraction separated. After its isolation by filtration and drying, 12.5 g of white lumpy powder with the melt, point of 141-144°C was obtained (this fraction contained 86.7% of the desired product, 12.7% of 2,3-dihydrophtalazine-l,4-dione, and the rest to 100 % were other unidentifiable impurities according to HPLC). The yield of the isolated amine calculated to the pure substance was 73%.
(S)-isomer 99,79 % (R)-isomer 0,21 % (S)-isomer 99,81 % / (R)-isomer 0,19 %
Crude rivaroxaban with the chemical purity of 99.5%, containing 0.21% of the (i?)-enantiomer and 0.50% of unidentified impurities, was crystallized from a mixture of acetic acid and ethanol.
Crystallization 1: 22 g of crude rivaroxaban were dissolved in 180 ml of acetic acid at boiling and the obtained solution was still hot filtered. The filtrate was brought to boil again and 400 ml of ethanol was gradually added to the boiling solution. The mixture was stirred under slow cooling for ca. 1 hour (resulting temperature of the suspension ca. 28°C). Subsequently, filtration was performed, the cake was washed with 2x30 ml of ethanol and vacuum-dried. 20.3 g of the product was obtained, melt, point 227.5-228.5°C. The yield of the crystallization was 92%, HPLC 99.8%, content of the ( ?)-enantiomer 0.21%, contents of unidentified impurities 0.15%.
Crystallization 2: 20 g of once crystallized rivaroxaban were dissolved in 150 ml of acetic acid at boiling and the obtained solution was still hot filtered (the filter was washed with 20 ml of boiling acetic acid to the filtrate). The filtrate was brought to boil again and 340 ml of ethanol was gradually added to the boiling solution. The mixture was stirred under slow cooling for ca. 1.5 hours (resulting temperature of the suspension ca. 26°C). Subsequently, filtration was performed, the cake was washed with 2x50 ml of ethanol and vacuum-dried. 19.1 g of the product was obtained, melt, point 230-231 °C. Crystallization yield 96%, HPLC 99.96%, content of the (fl)-enantiomer 0.19%, the contents of unidentified impurities was 0.04%.
After two crystallizations the final chemical purity of rivaroxaban achieved was 99.96%, the contents of unidentified impurities was reduced from 0.50% to 0.04%. After two crystallizations the achieved content of the (/?)-isomer was 0.19%, which is an excessive value (limit 0.15%) and is comparable to the initial level of 0.21%. The yield of rivaroxaban after the two crystallizations was 88% (calculated to the starting crude rivaroxaban).
| WO2001047919A1 | Dec 11, 2000 | Jul 5, 2001 | Bayer Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2004060887A1 | Dec 24, 2003 | Jul 22, 2004 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| WO2007116284A1 | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2009023233A1 | Aug 14, 2008 | Feb 19, 2009 | Concert Pharmaceuticals Inc | Substituted oxazolidinone derivatives |
| WO2010043110A1 | Oct 9, 2009 | Apr 22, 2010 | Changzhou Multiple Dimension Institute Of Industry Technology Co., Ltd. | A preparation method of high-purity l-carnitine |
| WO2010082627A1 | Jan 15, 2010 | Jul 22, 2010 | Daiso Co., Ltd. | Process for producing 2-hydroxymethylmorpholine salt |
| WO2010124835A1 | Apr 27, 2010 | Nov 4, 2010 | Belte Ag | Aluminium-silicon diecasting alloy for thin-walled structural components |
| WO2011080341A1 | Jan 3, 2011 | Jul 7, 2011 | Enantia, S.L. | Process for the preparation of rivaroxaban and intermediates thereof |
| WO2011098501A1 | Feb 10, 2011 | Aug 18, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2011102640A2 | Feb 16, 2011 | Aug 25, 2011 | Hanmi Holdings Co., Ltd. | Method for preparing sitagliptin and amine salt intermediates used therein |
| WO2012159992A1 * | May 18, 2012 | Nov 29, 2012 | Interquim, S.A. | Process for obtaining rivaroxaban and intermediate thereof |
| CN102786516A * | Aug 21, 2012 | Nov 21, 2012 | 湖南师范大学 | Method for synthesizing rivaroxaban |
| US7157456 | Dec 11, 2000 | Jan 2, 2007 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| US7816355 * | Apr 28, 2009 | Oct 19, 2010 | Apotex Pharmachem Inc | Processes for the preparation of rivaroxaban and intermediates thereof |
| US20110034465 | Feb 10, 2011 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
TAKE A TOUR
College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou, CHINA

Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang

DONGYANG


 .
. DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
{kind=link}
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus