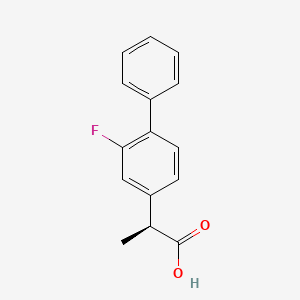
Cas 51543-39-6,
MW 244.26,
MF C15 H13 F O2
[1,1'-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (αS)-
- [1,1'-Biphenyl]-4-acetic acid, 2-fluoro-α-methyl-, (S)-
- (+)-(S)-Flurbiprofen
- (+)-Flurbiprofen
- (2S)-2-(2-Fluoro-1,1'-biphenyl-4-yl)propanoic acid
- (2S)-2-(2-Fluoro-4-biphenyl)propanoic acid
- (S)-Flurbiprofen
- Dexflurbiprofen
- Esflurbiprofen
- S-(+)-Flurbiprofen
- d-Flurbiprofen
On October 20, 2014, Taisho filed for manufacturing and marketing approval for TT-063 from the Ministry of Health, Labour and Welfare as a new drug candidate that will follow the Type 2 diabetes treatment Lusefi®, which was launched in May 2014. TT-063 is a patch formulation that has been co-developed by Taisho and TOKUHON Corporation with the aim of obtaining an indication for osteoarthritis. In Phase 3 clinical trials comparing TT-063 with therapeutic drugs already on the market, TT-063 has been found to be more effective than the control drugs in patients with osteoarthritis of the knee joint (January 16, 2014 announcement ).
Furthermore, Taisho is also preparing to file for approval from the Ministry of Health, Labour and Welfare for CT-064, an oral formulation of the osteoporosis treatment agent Bonviva launched in August 2013. Taisho has confirmed the effectiveness of CT-064 for osteoporosis patients through Phase 3 clinical trials (September 22, 2014 announcement).

In the central nervous system field, TS-091 transitioned from Phase 1 to Phase 2 in Japan in May 2014. Clinical trials of TS-091 have commenced to confirm the effectiveness of this drug in patients with central disorders of hypersomnolence. In addition, Phase 1 clinical trials of TS-091 have commenced overseas. TS-111 and TS-121 are undergoing Phase 1 clinical trials overseas with the aim of obtaining an indication for depression.
Faced with intensifying competition in new drug discovery, we will jointly implement R&D activities with research institutions outside the Taisho Group, and with companies in Japan and overseas, as we work to enhance our drug development pipeline (lineup of drugs in development). Our goal is to discover many more new drugs, primarily in our priority fields.
| Company | Taisho Pharmaceutical Holdings Co. Ltd. |
| Description | Topical anti-inflammatory analgesic patch containing S-flurbiprofen |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Standard Indication | Osteoarthritis |
| Indication Details | Treat osteoarthritis (OA) and scapulohumeral periarthritis |
| Regulatory Designation | |

Scheme 2.
Reagents and conditions: (a) THF, EDC, Et3N; (b) TFA; (c) 0.5 equiv 2,5-dimethoxybenzoquinone, EtOH, 50–80 °C for 3–5 h; (d) 1 equiv naphthoquinone, MeOH, rt, overnight.
...................................................
2-(6-methoxynaphthalen-2-yl) propanoic acid By way of illustration, chemically, flurbiprofen is 2-(2-fluoro-4-biphenylyl) propionic acid and is described in US Patent No. 3,755,427. NSAIDs, such as flurbiprofen, are usually supplied as a racemate. However, recently there has been renewed interest in the separate enantiomers of flurbiprofen, i.e. S-flurbiprofen and R-flurbiprofen.

R-Flurbιprofen

S-Flurtιprofen
Flurbiprofen is a potent inhibitor of cyclooxygenase (both COX-I and COX-2) in humans and it is understood that the inhibitory effect lies predominantly in the S- enantiomer.
Flurbiprofen is generally produced in the form of a racemic compound. It is known that from the racemic compound, flurbiprofen having a high optical purity can be produced by an optical resolution method using, for example, an optically active amine compound, such as α-phenylethylamine, as an optical resolution agent, as is described in US Patent No. 5,599,969. In addition, whether dealing with racemic, S- or R- 2-aryl propionic acid, there is also a need to make the synthetic process as efficient as possible.
Example 2 - Ibuprofen
Example 2.1 Resolution procedure
Racemic ibuprofen (530g) is dissolved in toluene (1335ml) and methanol (900ml).
The mixture is heated to dissolve the solid. S-1-Phenylethylamine (247g) is dissolved in toluene (200ml) and the solution is added with stirring at 600C over about 3 hours while the temperature is maintained at about 65-700C. The mixture is cooled gradually to 0 to 50C to induce crystallisation and stirred at this temperature for 1 hour. The crystals are filtered off, washed with toluene (600ml) and dried in a Vacuum oven at 550C to form crude S-ibuprofen / S-1-phenylethylamine salt (635g).
Crude S-ibuprofen / S-1-phenylethylamine salt (635g) is stirred with toluene (1930ml) and methanol (800ml) and the mixture is heated to 6O0C to dissolve the solid. The solution is cooled gradually to 0 to 5°C to induce crystallisation. The crystals are filtered off and dried in a vacuum oven at 55°C to form pure S-ibuprofen / S-I- phenylethylamine salt (510g). This recrystallisation of the S-ibuprofen / S-I- phenylethylamine salt may be repeated if necessary to upgrade the enantiomeric purity if required.
Pure S-ibuprofen / S-1-phenylethylamine salt (485g) is mixed with toluene (1700ml) with stirring. Water (300ml) and concentrated hydrochloric acid (17Og) are added and
÷ibe mixture is stirred at 600C. The lower aqueous layer is separated off and the upper organic layer is retained. The hydrochloric acid wash is repeated, then the toluene solution is washed with water. Water (370ml) and 47% sodium hydroxide
(118g) are added and the solution is heated to 600C and allowed to settle. The lower aqueous layer is separated and the upper toluene layer is washed with water. The aqueous phases are combined and heptane (420ml) is added. Hydrochloric acid
(130g) is added and the mixture is heated to 600C, stirred and settled. The organic layer is separated off and washed with water. The solution is cooled to -100C to induce crystallisation and the crystals are separated off by filtration, washed with heptane and dried under vacuum to yield (S)-ibuprofen (28Og) at an enantiomeric purity of over 99%.
Example 2.2 Racemisation procedure
Toluene/methanol mother liquors from the filtration of crude S-ibuprofen / S-I- phenylethylamine salt in the resolution procedure (2400ml, containing an estimated 130g of ibuprofen) is charged into a 3 L 3 necked round bottomed flask and methanol and toluene are distilled out at atmospheric pressure (volume removed approximately 1400 ml). The batch is then cooled to around 60°C and washed twice with hydrochloric acid (20 ml concentrated hydrochloric acid in 200 ml of water), and then twice with water (200 ml). Toluene is charged (80 ml) followed by methanol (200 ml) and caustic soda solution (45Og of 28% w/w solution, 5 molar equivalents). The mixture is heated to reflux for about 6 hours. Solvent is then removed at atmospheric pressure until the vapour temperature reaches approximately 85°C. The mixture is cooled to around 60°C and concentrated hydrochloric acid is charged at about 60 to 70°C until the pH of the mixture is 1 or less. The layers are allowed to separate and the bottom aqueous layer removed. The organic layer is washed with water (200 ml) and then azeotroped to dryness using a Dean and Stark trap. A solution of racemic ibuprofen in toluene remains.
................................................
PATENT
Preparation of R - (+) _ flurbiprofen:
The racemic flurbiprofen as a starting material, to obtain an intermediate product of formula I as shown and then the ester prepared as shown in Formula II with 5-isosorbide monobenzyl ether, ester hydrolysis after obtained R - (+) - flurbiprofen;

wherein, in formula I, X is Cl or Br;
(2) by the R - (+) _ flurbiprofen obtained (RS) - flurbiprofen:
The R _ (+) _ flurbiprofen 200mg, potassium hydroxide 150mg, 0. 5mL water into IOmL reaction flask and heated to 120 ° C and held for 2h, then water was added 15mL, cooled to room temperature, the resulting stirring the mixed solution with 10% hydrochloric acid to pH = 0. 5, extracted with ethyl acetate, combined several layers, washed with water until neutral, the organic solvent is recovered, the resulting residue was added at 60~90 ° C under an appropriate amount of petroleum ether by recrystallization, obtained (RS) - flurbiprofen 100mg, 50% yield.
(3) Preparation of (S) -⑴- flurbiprofen:
In 25mL single-necked flask, followed by adding (RS) - flurbiprofen 123mg, Portugal TOA 29. 8mg, isopropanol lmL, the mixture was stirred at reflux until clear, half the amount of the solvent evaporated under reduced pressure except , set the refrigerator overnight. The precipitate was collected by suction filtration as white crystals, after washing a small amount of isopropanol, which was dissolved in water, washed with 10% aqueous sodium hydroxide (10% NaOH mean mass fraction) adjusted pH = 13, the sheet-like precipitate was filtered off Portuguese octylamine white crystals. The resulting filtrate was added dropwise with stirring 10% hydrochloric acid to pH = 1, extracted with ethyl acetate, the organic layer was washed with water to recover the solvent, the resulting residue was purified by an appropriate amount of petroleum ether and recrystallized at 60~90 ° C. The product was collected by filtration, and dried in vacuo to give a white (S) - (+) _ flurbiprofen needle crystal 45. 3mg, 65% yield, mp 102~103 ° C, [α] = + 44 ° (C = 1, methanol), ee value of 92.6% (ee value measurement method: (S) - (+) - flurbiprofen after chiral amine derivatization reagents, by HPLC analysis).
wherein in step (3) is a byproduct eleven R _ (+) _ flurbiprofen, its follow step (1) of racemic reused.
Step (1) of the specific operation is as follows:
(la) 1:. Synthesis of 2,6-sorbitol dehydration -D- -5- benzyl ether: 4: 3
250ml volumetric flask isosorbide 18. 25g (125mmol), lithium hydroxide monohydrate 5. 25g (125mmol) and 60ml of dimethyl sulfoxide (DMSO), heated to 90 ° C, stirred for 30min, constant pressure equalizing dropping funnel was added dropwise benzyl chloride 14. 4ml (125mmol), 90 ° C the reaction 19-20h, reaction mixture was adjusted to pH 1 with 2M hydrochloric acid, extracted with ethyl acetate (50ml * 3), the organic layers combined, washed with water ( 30ml * 2), dried over anhydrous sodium sulfate overnight, filtered and concentrated residue Cheng baby gel column chromatography (petroleum ether: ethyl acetate = 5: 1) to give a cream solid, that is 1: 4: 3: 2,6 Dehydration -D- sorbitol -5- benzyl ether 24. 5g, m.p. 59 ~61 ° C.
(Ib) · 2- (2- fluoro-4-biphenylyl) propionyl chloride Synthesis
50ml vial before racemic flurbiprofen was added 2. 44g (IOmmol), anhydrous toluene 20ml, freshly distilled thionyl chloride was added dropwise 0. 8ml (Ilmmol), N, N- dimethylformamide amide (DMF) 2 dropwise, stirred at room temperature 2h, the solvent was distilled off under reduced pressure to give a pale yellow gum, i.e., 2- (2-fluoro-4-biphenylyl) propionyl chloride, it was used directly in the reaction without isolation.
(lc). R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester synthesis
The (Ib) resulting acid chloride was dissolved in 20ml of dry toluene was added dropwise at room temperature, dimethyl amine 3. 5ml, solid precipitation, stirred for about Ih, ice salt bath, a bath temperature of minus 10-15Ό, stirred at this temperature IOmin so, and then the constant pressure dropping funnel (Ia) 5 isosorbide monobenzyl ether (2. 83g, 12mmol) in toluene, keeping the reaction temperature, stirring 8h. The ice bath was removed and the reaction mixture under reduced pressure to remove the solvent, the residue was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous sodium sulfate overnight, ethyl acetate was removed under reduced pressure, the residue was a white gel, recrystallized from petroleum ether to give a white solid that R-2- (2- fluoro-4-biphenylyl) propionic acid 5- isosorbide monobenzyl ether ester 3. 65g (7. 88mmol), in order to put the racemic flurbiprofen yield based on 78.8%.
(ld) R - Synthesis of flurbiprofen - (+)
Under ice bath (Ic) obtained R-2- (2- fluoro-4-biphenylyl) propionic acid monobenzyl ether isosorbide 5- ester 2. 3Ig (5mmol) was dissolved in 20ml of acetone / water (1/1) was added Iml hydrochloric acid to adjust pH to 3, stirred for 3-4h, the reaction solution was extracted with ethyl acetate (20ml * 2), sash organic layer was washed with ice (10ml * 2), dried over anhydrous sodium sulfate overnight , filtration, and the filtrate was concentrated, the residue was recrystallized from ether to give white crystals, i.e. L-flurbiprofen 1.02g (4 18mmol.), yield 83.5%, optical purity 93% (HPLC method); input-racemic flurbiprofen dollars, the total yield of 78.8% * 83.5% = 65.8%.
Step (1) reaction of the formula:

FLURBIPROFEN RACEMIC
3-Fluoro-4-phenyl-α-methylphenylacetic acid 1
M.p. 110-113°C (lit.3d 111-113.5°C).
1 H NMR (CDCl3, δ ppm) 7.51-7.55 (m, 2H), 7.49-7.37 (m, 4H), 7.21-7.16 (m, 2H), 3.85-3.78 (q, 1H, J = 7.1 Hz, CH), 1.60-1.57 (d, 3H, J = 7.1 Hz, CH3);
13C NMR (CDCl3 δ ppm) 180.4 (COOH), 161.3 & 158.0 (3-Ar-C), 140.9 & 140.8, 135.4, 130.9 & 130.8 (5-Ar-C), 128.9, 128.4, 128.2 & 128.0 (4-Ar-C), 127.7 (4′-Ar-C), 123.7 & 123.7 (6-Ar-C), 115.5 & 115.2 (2-Ar-C), 44.8 (CH), 18.0 (CH3).
(d) Sagami Chemical Research Center. Jpn. Kokai Tokkyo Koho JP 8216840, 1982 (Chem. Abstr. 1982, 97: 5996s).

 |
| RACEMIC |
Flurbiprofen
CAS : 5104-49-4
: 2-Fluoro-a-methyl[1,1¢-biphenyl]-4-acetic acid
Additional Names: 2-(2-fluoro-4-biphenylyl)propionic acid; 3-fluoro-4-phenylhydratropic acid
Manufacturers' Codes: BTS-18322; U-27182
Trademarks: Adfeed (Lead Chem.); Ansaid (Pfizer); Antadys (Thžamex); Cebutid (Boots); Froben (Boots); Flurofen (Boots); Ocufen (Allergan); Stayban (Boots); Zepolas (Mikasa)
Molecular Formula: C15H13FO2
Molecular Weight: 244.26
Percent Composition: C 73.76%, H 5.36%, F 7.78%, O 13.10%
Literature References: Prepn: FR M5737; Adams et al., US 3755427 (1968, 1973 both to Boots Co., Ltd.). Pharmacology: Chalmers et al., Ann. Rheum. Dis. 31, 319 (1972); ibid. 32, 58 (1973); Glenn et al., Agents Actions 3, 210 (1973); Nishizawa et al.,Thromb. Res. 3, 577 (1973). HPLC determn in urine and plasma: J. M. Hutzler et al., J. Chromatogr. B 749, 119 (2000). Symposium on pharmacokinetics and clinical efficacy in pain management: Am. J. Med. 80, Suppl. 3A, 1-157 (1986).
Properties: Crystals from petr ether, mp 110-111°. Slightly sol in water (pH 7.0); readily sol in most polar solvents.
Melting point: mp 110-111°
Therap-Cat: Anti-inflammatory; analgesic.
|


| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Methods to accelerate the isolation of novel cell strains from pluripotent stem cells and cells obtained thereby [US2008070303] | 2006-11-21 | 2008-03-20 |
| Herpes Virus-Based Compositions and Methods of Use in the Prenatal and Perinatal Periods [US2008226601] | 2006-06-05 | 2008-09-18 |
| METHOD OF REDUCING ABETA42 AND TREATING DISEASES [US2008021085] | 2007-06-21 | 2008-01-24 |
| METHODS TO ACCELERATE THE ISOLATION OF NOVEL CELL STRAINS FROM PLURIPOTENT STEM CELLS AND CELLS OBTAINED THEREBY [US2010184033] | 2009-07-16 | 2010-07-22 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US2011112064] | 2011-05-12 | |
| PROCESS FOR THE MANUFACTURE OF RACEMIC 2-ARYL-PROPIONIC ACID [US2011172460] |
| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Nitroxyderivatives having antinflammatory, analgesic and antithrombotic activity [US6613784] | 2003-09-02 | |
| Global method for mapping property spaces [US6675136] | 2004-01-06 | |
| Method of reducing Abeta42 and treating diseases [US2006004086] | 2006-01-05 | |
| 11-Beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia [US7179802] | 2004-06-03 | 2007-02-20 |
| 11-BETA-HYDROXYSTEROID DEHYDROGENASE 1 INHIBITORS USEFUL FOR THE TREATMENT OF DIABETES, OBESITY AND DYSLIPIDEMIA [US6730690] | 2004-03-11 | 2004-05-04 |
| Process for producing optically active flurbiprofen [US7214820] | 2006-06-22 | 2007-05-08 |
| Pyridyl Amide T-Type Calcium Channel Antagonists [US7875636] | 2009-11-05 | 2011-01-25 |
| METHOD FOR PRODUCING OPTICALLY ACTIVE ESTER AND METHOD FOR PRODUCING OPTICALLY ACTIVE CARBOXYLIC ACID [US8115008] | 2010-09-16 | 2012-02-14 |
| DRUG SUBSTANCE PREPARATIONS, PHARMACEUTICAL COMPOSITIONS AND DOSAGE FORMS [US2010087538] | 2010-04-08 | |
| (R)-2-(3-Benzoylphenyl)propionic acid salts and pharmaceutical preparations containing them [EP0935961] | 1999-08-18 | 2008-04-02 |
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

Taisho Pharmaceutical Co., Ltd. (大正製薬株式会社 Taishō Seiyaku Kabushiki-gaisha) (TYO: 4535) is a Japanese pharmaceutical company based in Tokyo.

////////////
TIRUPATI, INDIA
 .
.
| TIRUPATI తిరుపతి | |
|---|---|
| City | |

Clockwise from top: Tirumala Venkateswara Temple, Tirumala ghat road, City skyline and Chandragiri fort
| |

Tirupati
Location in Andhra Pradesh, India
| |
| Coordinates: 13.65°N 79.42°ECoordinates: 13.65°N 79.42°E | |
| COUNTRY | India |
| STATE | Andhra Pradesh |
| REGION | Rayalaseema |
| DISTRICT | Chittoor |
| GOVERNMENT | |
| • MEMBER OF PARLIAMENT | Varaprasad Rao Velagapalli |
| AREA | |
| • CITY | 24 km2 (9 sq mi) |
| ELEVATION | 161 m (528 ft) |
| POPULATION (2011)[1] | |
| • CITY | 287,035 |
| • DENSITY | 12,000/km2 (31,000/sq mi) |
| • METRO[2] | 459,985 |
| LANGUAGES | |
| • OFFICIAL | Telugu |
| TIME ZONE | IST (UTC+5:30) |
| PIN | 517501 |
| TELEPHONE CODE | +91–877 |
| VEHICLE REGISTRATION | AP 03 |
| WEBSITE | Tirupati Mucnicipal Corporation |

 .
. .
.Kapila Theertham in Tirupati








Food Service During Tirumala Tirupati Devastanam's 'Srinivasa Kalyanam Utsavam' at MARG Swarnabhoomi


