Olopatadine hydrochloride
Cis form, Z Isomer
( Z ) - 1 1 - [ 3 - ( D i m e t h y l a m i n o ) p r opy l i d e n e ] - 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride
ALO 4943A; Allelock; KW 4679; Opatanol; Patanol;
| (11Z)-11-[3-(Dimethylamino)propylidene]-6,11-dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride; | |
| CAS Number: | 140462-76-6 |
| unii 2XG66W44KF | |
| Molecular form.: | C₂₁H₂₄ClNO₃ |
| Appearance: | White Solid |
| Melting Point: | >240˚C (dec.) |
| Mol. Weight: | 373.87 |
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT,
SPECTROSCOPY FROM NET
( Z ) - 1 1 - [ 3 - ( D i m e t h y l a m i n o ) p r opy l i d e n e ] - 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride.
Cis-olopatadinehydrochloride /olopatadinehydrochloride
mp 231−233 °C (dec);
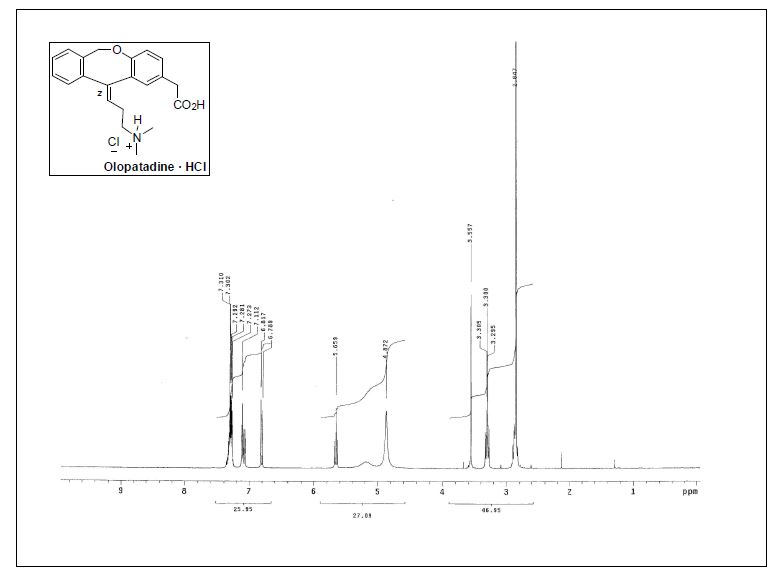
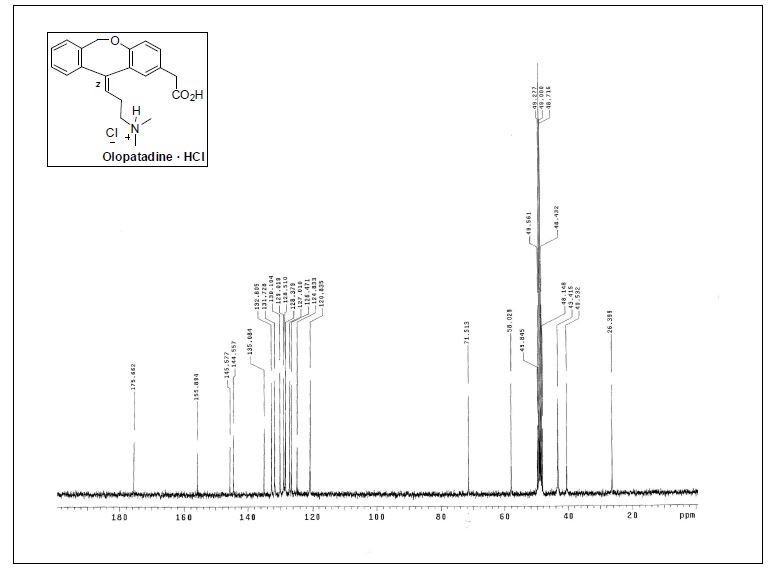
1H NMR (300 MHz, CD3OD) δ 2.86 (s, 6H), 2.83−2.91 (m, 2H), 3.28−3.34 (m, 2H), 3.57 (s, 2H), 5.19 (br, 2H), 5.67 (t,J = 7.3 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 7.07−7.13 (m, 2H), 7.26−7.37 (m, 4H);
13C NMR (75.4 MHz, CD3OD) δ 26.4 (CH2), 40.5(CH2), 43.4 (2CH3), 58.0 (CH2), 71.5 (CH2), 120.3 (CH), 124.8 (C),126.5 (CH), 127.0 (CH), 128.4 (C), 128.5 (CH), 129.0 (CH), 130.1(CH), 131.7 (CH), 132.8 (CH), 135.1 (C), 144.5 (C), 145.6 (C),155.9 (C), 175.7 (C);
IR (KBr) 1225, 1491, 1716, 2927 cm−1.
Anal.Calcd for C21H24NClO3·H
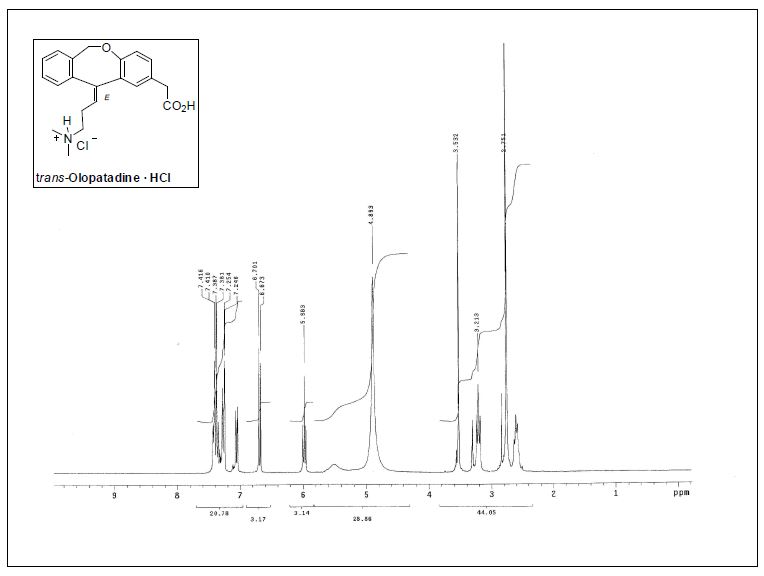
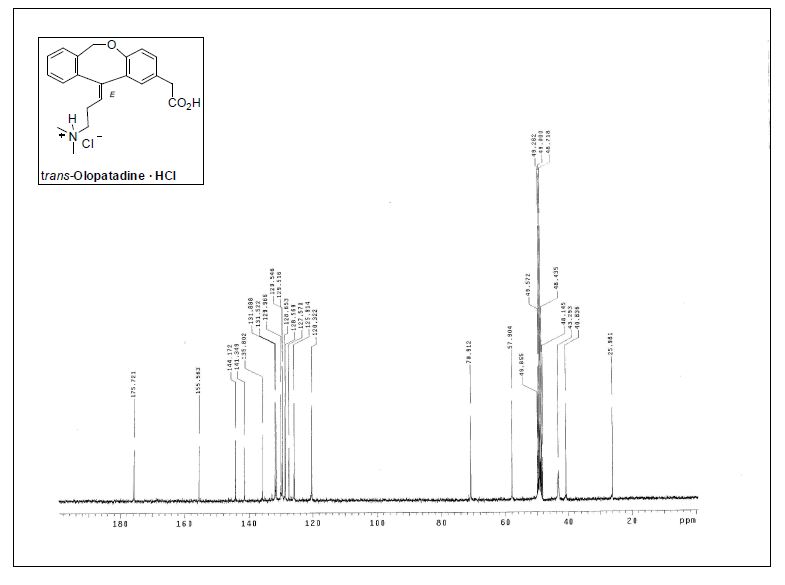
( E ) - 1 1 - [ 3 - ( D i m e thy l ami n o ) p r o p y l i d e n e ] - 6 , 1 1 -dihydrodibenz[b,e]oxepin-2-acetic Acid Hydrochloride.
trans-olopatadinehydrochloride
mp 170−173 °C;
1H NMR (300 MHz, CD3OD) δ 2.56−2.63 (m, 2H), 2.75 (s,6H), 3.13 (t, J = 7.6 Hz, 2H), 3.53 (s, 2H), 4.78 (br, 1H), 5.51 (br,
1H), 5.98 (t, J = 7.2 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 7.06 (dd, J =8.3, 2.3 Hz, 1H), 7.25−7.44 (m, 5H);
1H), 5.98 (t, J = 7.2 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 7.06 (dd, J =8.3, 2.3 Hz, 1H), 7.25−7.44 (m, 5H);
13C NMR (75.4 MHz, CD3OD)δ 26.0 (CH2), 40.8 (CH2), 43.3 (2CH3), 57.9 (CH2), 70.9 (CH2),120.3 (CH), 125.9 (CH), 127.6 (C), 128.5 (C), 128.6 (CH), 129.5(2CH), 130.0 (CH), 131.5 (CH), 132.0 (CH), 135.8 (C), 141.3 (C),144.2 (C), 155.6 (C), 175.7 (C);
IR (KBr) 1223, 1484, 1725, 2960cm−1.
Anal. Calcd for C21H24NClO3·H2O: C, 64.36; H, 6.69; N, 3.57.
Found: C, 64.66; H, 6.47; N, 3.56.
Found: C, 64.66; H, 6.47; N, 3.56.
http://pubs.acs.org/doi/full/10.1021/jo300925c
J. Org. Chem., 2012, 77 (14), pp 6340–6344
DOI: 10.1021/jo300925c
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT,
PATENT
https://www.google.com/patents/WO2007110761A2?cl=en


 Scheme 2:
Scheme 2:




 Scheme 3:
Scheme 3:



 A significant disadvantage of the synthetic route depicted in Scheme 4 is the diastereoselectivity of the dehydration step, which gives up to 90% of the undesired E-isomer. The last step (oxidation) is not described in this publication.
A significant disadvantage of the synthetic route depicted in Scheme 4 is the diastereoselectivity of the dehydration step, which gives up to 90% of the undesired E-isomer. The last step (oxidation) is not described in this publication.



 acid
acid


 acid
acid




(Z)-I l-[3-Dimethylamino- Olo-HCl propylidene]-6,l 1-dihydro- dibenz[b,e]oxepin-2-acetic acid hydrochloride

PATENT
https://www.google.com/patents/WO2007110761A2?cl=en
Olopatadine-HCl ([(Z)-3-(dimethylamino)propylidene]-6,ll-dihydrodibenz[b,e]oxepin-2- acetic acid hydrochloride) is a selective histamine Hl -receptor antagonist that is used for the treatment of ocular symptoms of seasonal allergic conjunctivitis. The compound may be administered in a solid oral dosage form or as an ophthalmic solution.
Olopatadine-HCI
[(Z)-3-(Dimethylamino)propylidene]-6,11-dihydro- dibenz[b,e]oxepin-2-acetic acid hydrochloride
Olopatadine is stated to be an effective treatment for symptoms of allergic rhinitis and urticaria (e.g., sneezing, nasal discharge and nasal congestion), as well as in the treatment of various skin diseases, such as eczema and dermatitis.
Olopatadine and its pharmaceutically acceptable salts are disclosed in EP 0214779, U.S. Patent No. 4,871,865, EP 0235796 and U.S. Patent No. 5,116,863. There are two general routes for the preparation of olopatadine which are described in EP 0214779: One involves a Wittig reaction and the other involves a Grignard reaction followed by a dehydration step. A detailed description of the syntheses of olopatadine and its salts is also disclosed in Ohshima, E., et al., J. Med Chem. 1992, 35, 2074-2084. EP 0235796 describes a preparation of olopatadine derivatives starting from 1 l-oxo-6,11- dihydroxydibenz[b,e]oxepin-2-acetic acid, as well as the following three different synthetic routes for the preparation of corresponding dimethylaminopropyliden-dibenz[b,e]oxepin derivatives, as shown in schemes 1-3 below:
Scheme 1:
HaIMgCH2CH2CH2NMe2
R1OH or
R2CI
R1 = R2 = alkyl group R1 = H, R2 = trityl group
HaIMgCH2CH2CH2NMe2
Ph3P Hal' sHal
R3 = COOH, etc.
The syntheses of several corresponding tricyclic derivatives are disclosed in the same manner in EP 0214779, in which the Grignard addition (analogous to Scheme 1) and the Wittig reaction (analogous to Scheme 3) are described as key reactions.
The synthetic routes shown above in Schemes 2 and 3 for the preparation of olopatadine are also described in Ohshima, E., et al., J Med. Chem. 1992, 35, 2074-2084 (schemes 4 and 5 below). In contrast to the above-identified patents, this publication describes the separation of the Z/E diastereomers (scheme 5). Scheme 4:
65% Ph3CCI
81% CIMgCH2CH2CH2NMe2
Scheme 5 below depicts a prior art method disclosed in Ohshima, E., et al., supra.
Scheme 5:
Each of the prior art methods for synthesis of olopatadine have significant cost and feasibility disadvantages. Specifically with the respect to the method set forth in Scheme 5, the disadvantages include: (1) the need for excess reagents, e.g. 4.9 equivalents Wittig reagent and 7.6 equivalents of BuLi as the base for the Wittig reaction, which can be expensive;
(2) the need to use Wittig reagent in its hydrobromide salt form, so that additional amounts of the expensive and dangerous butyllithium reagent are necessary for the "neutralization" of the salt (i.e., excess butyllithium is required because of the neutralization);
(3) because 7.6 equivalents of the butlylithium are used (compared to 9.8 equivalents of the (Olo-IM4) Wittig reagent), the Wittig reagent is not converted completely to the reactive ylide form, and thus more than 2 equivalents of the Wittig reagent are wasted;
(4) the need for an additional esterifϊcation reaction after the Wittig reaction (presumably to facilitate isolation of the product from the reaction mixture) and the purification of the resulting oil by chromatography;
(5) the need to saponify the ester and to desalinate the reaction product (a diastereomeric mixture) with ion exchange resin, prior to separating the diastereomers;
(6) the need, after the separation of the diastereomers, and liberation of the desired diastereomer from its corresponding pTsOH salt, to desalinate the product (olopatadine) again with ion exchange resin;
(7) the formation of olopatadine hydrochloride from olopatadine is carried out using 8 N HCl in 2-propanol, which may esterify olopatadine and give rise to additional impurities and/or loss of olopatadine; and
(8) the overall yield of the olopatadine, including the separation of the diastereomers, is only approximately 24%, and the volume yield is less than 1%.
As noted above, the known methods for preparing olopatadine in a Wittig reaction use the intermediate compounds 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid and 3- dimethylaminopropyltriphenylphosphonium bromide hydrobromide. Preparation of these chemical intermediates by prior art syntheses present a number of drawbacks that add to the cost and complexity of synthesizing olopatadine.
One known method for preparation of the compound 6,11-dihydro-l 1-oxo- dibenz[b,e]oxepin-2-acetic acid is depicted in Scheme 6, below. See also, U.S. Patent No. 4,585,788; German patent publications DE 2716230, DE 2435613, DE 2442060, DE 2600768; Aultz, D.E., et al., J Med. Chem. (1977), 20(1), 66-70; and Aultz, D.E., et al., J Med. Chem. (1977), 20(11), 1499-1501. Scheme 6:
COOE
In addition, U.S. Patent No. 4,417,063 describes another method for the preparation of 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid, which is shown in Scheme 7. Scheme 7:
Ueno, K., et al., J Med. Chem. (1976), 19(7), 941, describes yet another prior art method for preparing 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid, which is shown below in Scheme 8. Scheme 8:
Further, as depicted in Scheme 9, below, U.S. Patent Nos. 4,118,401; 4,175,209; and 4, 160,781 disclose another method for the synthesis of 6, 11 -dihydro- 11 -oxo-dibenz[b,e]oxepin-2- acetic acid.
Scheme 9:
AICI3
6,11 -dihydro-11 -oxo-dibenz- [b,e]oxepin-2-acetic acid
JP 07002733 also describes the preparation of 6,11 -dihydro- 1 l-oxo-dibenz[b,e]oxepin-2- acetic acid, as follows in Scheme 10, below.
Scheme 10:
Specific methods and reagents for performing the intramolecular Friedel-Crafts reaction for cyclizing 4-(2-carboxybenzyloxy)-phenylacetic acid to form 6,11 -dihydro-11-oxo- dibenz[b,e]oxepin-2-acetic acid are described in (1) EP 0068370 and DE 3125374 (cyclizations were carried out at reflux with acetyl chloride or acetic anhydride in the presence of phosphoric acid, in toluene, xylene or acetic anhydride as solvent); (2) EP 0069810 and US 4282365 (cyclizations were carried out at 70-80° C with trifluoroacetic anhydride in a pressure bottle); and (3) EP 0235796; US 5,116,863 (cyclizations were carried out with trifluoroacetic anhydride in the presence of BF3 »OEt2 and in methylene chloride as solvent).
Turning to the Wittig reagent for use in preparing olopatadine, 3- dimethylaminopropyltriphenylphosphonium bromide-hydrobromide and methods for its preparation are described in U.S. Patent Nos. 3,354,155; 3,509,175; 5,116,863, and EP 0235796, and depicted in Scheme 11 below. Scheme 11:
Corey, E. J., et al, Tetrahedron Letters, Vol. 26, No. 47, 5747-5748, 1985 describes a synthetic method for the preparation of 3-dimethylaminopropyltriphenylphosphonium bromide (free base), which is shown below in Scheme 12. Scheme 12:
The prior art methods for preparing olopatadine and the chemical intermediates 6,11- dihydro-ll-oxo-dibenz[b,e]oxepin-2-acetic acid, and 3- dimethylaminopropyltriphenylphosphonium bromide-hydrobromide (and its corresponding free base) are not desirable for synthesis of olopatadine on a commercial scale. For example, due to high reaction temperatures and the absence of solvents, the synthesis described in Ueno, K., et al., J. Med. Chem. (1976), 19(7), 941 and in U.S. Patent No. 4,282,365 for preparation of the intermediate 4-(2-carboxybenzyloxy)phenylacetic acid is undesirable for a commercial scale process, although the synthesis described in JP 07002733, and set forth in Scheme 13 below, is carried out in an acceptable solvent. Scheme 13:
OIO-1M1
The processes described in the literature for the intramolecular Friedel-Crafts acylation used to prepare 6,11-dihydro-l l-oxo-dibenz[b,e]oxepin-2-acetic acid are undesirable for commercial scale synthesis because they generally require either drastic conditions in the high boiling solvents (e.g. sulfolane) or they require a two step synthesis with the corresponding acid chlorides as intermediate. Furthermore the procedures for synthesizing 6,11-dihydro-l 1-oxo- dibenz[b,e]oxepin-2-acetic acid as set forth in European patent documents EP 0069810 and EP 0235796 use excess trifluoroacetic anhydride (see Scheme 14), and are carried out without solvent in a pressure bottle at 70-80° C (EP 0069810) or at room temperature in methylene chloride using catalytic amounts of BF3^Et2O (EP 0235796). Scheme 14:
According to the teachings in EP 0235795, a suspension of 3- bromopropyltriphenylphosphonium bromide (Olo-IM4) in ethanol was reacted with 13.5 equivalents of an aqueous dimethylamine solution (50%) to provide dimethylaminopropyltriphenylphosphonium bromide HBr. After this reaction, the solvent was distilled off and the residue was recrystallized (yield: 59%).
U.S. Patent No. 3,354,155 describes a reaction of 3-bromopropyltriphenylphosponium bromide with 4.5 equivalents dimethylamine. The solution was concentrated and the residue was suspended in ethanol, evaporated and taken up in ethanol again. Gaseous hydrogen bromide was passed into the solution until the mixture was acidic. After filtration, the solution was concentrated, whereupon the product crystallized (yield of crude product: 85%). The crude product was recrystallized from ethanol. A significant disadvantage of the prior art processes for making 3- dimethylaminopropyltriphenylphosphonium bromide hydrobromide involves the need for time consuming steps to remove excess dimethylamine, because such excess dimethylamine prevents crystallization of the reaction product. Thus, to obtain crystallization, the prior art processes require, for example, repeated evaporation of the reaction mixture (until dryness), which is undesirable for a commercial scale synthesis of olopatadine.
Corey, EJ., et al., Tetrahedron Letters, Vol. 26, No. 47, 5747-5748 (1985) describes the preparation of 3-dimethylaminopropyltriphenylphosphonium bromide (free base) from its corresponding hydrobromide salt. But the preparation of the free base, which uses an extraction step with methylene chloride as the solvent, is undesirable for commercial production because of the poor solubility of the free base in many of the organic solvents that are desirable for commercial production of chemical products, and because of the high solubility of the free base in water, causing low volume yields and loss of material. Furthermore according to this publication, the work up procedure gave an oil, which crystallized only after repeated evaporation in toluene.
It would be desirable to provide processes for preparing olopatadine on a large scale, e.g., on a commercial scale, in a manner that is cost efficient and provides olopatadine that has a low level of impurities, including a low level of the undesired diastereomer.
It further would be desirable to eliminate the need to derivatize the olopatadine product of the Wittig reaction, e.g., by esterification, in order to separate the olopatadine from the reaction mixture. It would be especially desirable to provide a method for preparing olopatadine that allows for isolation of olopatadine directly from the reaction mixture.
It would also be desirable to eliminate the need for the costly and dangerous base, butyllithium, that is used in previously described Wittig reactions for making olopatadine.
It would also be desirable to provide improved methods for preparing chemical intermediates used in the synthesis of olopatadine via a Wittig reaction.
In the description of the various aspects of applicants' invention that follows, reference may be made to the chemical intermediates, final products and byproducts in accordance with the nomenclature set forth immediately below.
C21H24CINO3
Exact Mass: 373.14
MoI. Wt.: 373.87
Scheme 15:

+ HBr extraction at pH = 42-46 with THF or MeTHF/iPrOH or BuOH

Scheme 16:

Scheme 17:

Olo-IM4ylide OIO-IM4 BP1

Olo-IM4 (free base OIO-1M4 BP1


1. WO2007110761A2.

1. WO2010007056A1.

1. WO2011033532A1.

| EP0351887A1 * | Aug 15, 1986 | Jan 24, 1990 | The Wellcome Foundation Limited | Tricyclic aromatic compounds |
| US3354155 * | Oct 12, 1964 | Nov 21, 1967 | Pfizer & Co C | Aminoalkylphosphonium compounds and process for the use thereof |
| US3681337 * | Dec 31, 1970 | Aug 1, 1972 | Ciba Geigy Corp | SUPPRESSION OF TRIS(ALKYLAMINO)-s-TRIAZINE FORMATION IN THE PRODUCTION OF CHLORO-BIS (ALKYLAMINO)-s-TRIAZINES THROUGH THE USE OF ADDITIONAL CYANURIC CHLORIDE |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | COREY, E.J. ET AL.: TETRAHEDRON LETTERS, vol. 26, no. 47, 1985, pages 5747-5748, XP002448423 cited in the application |
| 2 | * | LIU Y ET AL: "Synthesis of a new H1,receptor antgonist olopatadine" ZHONGGUO XIN YAO ZAZHI - CHINESE NEW DRUGS JOURNAL, GAI-KAN BIANJIBU, BEIJING, CN, vol. 15, no. 23, 2006, pages 2045-2046, XP008082683 ISSN: 1003-3734 |
| 3 | * | OHSHIMA E ET AL: "SYNTHESIS AND ANTIALLERGIC ACTIVITY OF 11-(AMINOALKYLIDENE)-6,11-DIHY DRODIBENZÚB,EOXEPIN DERIVATIVES" JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 35, no. 11, 1 May 1992 (1992-05-01), pages 2074-2084, XP000615220 ISSN: 0022-2623 cited in the application |
| 4 | * | XUE ET AL: "Study on the synthetic process of a novel anti-allergic agent olopatadine hydrochloride" ZHONGGUO YAOWU HUAXUE ZAZHI - CHINESE JOURNAL OF MEDICINAL CHEMISTRY, GAI-KAI BIANJIBU, SHENYANG, CN, vol. 14, no. 6, 2004, pages 363-364,367, XP008082682 ISSN: 1005-0108 |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2007119120A2 * | Dec 22, 2006 | Oct 25, 2007 | Medichem, S.A. | Crystalline polymorphic forms of olopatadine hydrochloride and processes for their preparation |
| WO2007119120A3 * | Dec 22, 2006 | Feb 14, 2008 | Monica Benito | Crystalline polymorphic forms of olopatadine hydrochloride and processes for their preparation |
| WO2009048086A1 * | Oct 2, 2008 | Apr 16, 2009 | Sumitomo Chemical Company, Limited | Method for purification of dibenzoxepin compound |
| WO2009054298A1 * | Oct 15, 2008 | Apr 30, 2009 | Sumitomo Chemical Company, Limited | Process for production of crystal of dibenzoxepin compound |
| WO2010089268A2 | Feb 1, 2010 | Aug 12, 2010 | Zach System S.P.A. | Process for preparing olopatadine and/or a pharmaceutically acceptable salt thereof |
| WO2010089268A3 * | Feb 1, 2010 | Oct 14, 2010 | Zach System S.P.A. | Process for preparing olopatadine and/or a pharmaceutically acceptable salt thereof |
| WO2010121877A3 * | Mar 23, 2010 | Mar 10, 2011 | F.I.S. Fabbrica Italiana Sintetici S.P.A. | Process for the preparation of olopatadine |
| WO2011027322A1 * | Sep 2, 2010 | Mar 10, 2011 | Ranbaxy Laboratories Limited | Extended release dosage form containing olopatadine for oral administration |
| WO2015063579A1 * | Nov 4, 2014 | May 7, 2015 | Laboratorio Chimico Internazionale S.P.A. | A process for the preparation of olopatadine and sylil intermediates thereof |
| CN102757339A * | Aug 1, 2012 | Oct 31, 2012 | 北京联本医药化学技术有限公司 | Improved preparation method of 4-(2-carboxybenzyloxy) phenylacetic acid |
| CN104262318A * | Sep 3, 2014 | Jan 7, 2015 | 石家庄创建医药科技有限公司 | Method for preparing olopatadine hydrochloride |
| EP2228371A1 * | Dec 22, 2006 | Sep 15, 2010 | Medichem, S.A. | New process for preparing olopatadine free base and/or its hydrochloride salt |
| EP2385045A1 * | Jan 21, 2010 | Nov 9, 2011 | Sumitomo Chemical Company, Limited | Process for producing dibenzoxepin compound |
| US8598352 | May 21, 2010 | Dec 3, 2013 | H. Lundbeck A/S | Preparation of nalmefene hydrochloride from naltrexone |
| US8835655 | Feb 1, 2010 | Sep 16, 2014 | Zach System S.P.A | Process for preparing olopatadine and/or a pharmaceutically acceptable salt thereof |
| US8871953 | Mar 23, 2010 | Oct 28, 2014 | F.I.S. Fabbrica Italiana Sintetiei S.p.A. | Process for the preparation of olopatadine |
| US8980939 | Jul 12, 2013 | Mar 17, 2015 | F.I.S. Fabbrica Italiana Sintetici S.P.A. | Process for the preparation of olopatadine |
| US20130273619 * | Apr 16, 2013 | Oct 17, 2013 | Basf Se | Process for the Preparation of (3E, 7E)-Homofarnesol |
Title: Olopatadine
CAS Registry Number: 113806-05-6
CAS Name: (11Z)-11-[3-(Dimethylamino)propylidene]-6,11-dihydrodibenz[b,e]oxepin-2-acetic acid
Molecular Formula: C21H23NO3
Molecular Weight: 337.41
Percent Composition: C 74.75%, H 6.87%, N 4.15%, O 14.23%
Literature References: Dual acting histamine H1-receptor antagonist and mast cell stabilizer. Prepn: E. Oshima et al., EP235796; eidem, US 5116863 (1987, 1992 both to Kyowa); eidem, J. Med. Chem. 35, 2074 (1992). Pharmacology: C. Kamei et al.,Arzneim.-Forsch. 45, 1005 (1995); J. M. Yanni et al., J. Ocul. Pharmacol. Ther. 12, 389 (1996). Receptor binding profile: N. A. Sharif et al., J. Pharmacol. Exp. Ther. 278, 1252 (1996). Clinical trial in allergic conjunctivitis: M. B. Abelson, L. Spitalny, Am. J. Ophthalmol. 125, 797 (1998).
Properties: Crystallized as the hemihydrate from 2-propanol + water, mp 188-189.5°.
Melting point: mp 188-189.5°
Derivative Type: Hydrochloride
CAS Registry Number: 140462-76-6
Manufacturers' Codes: AL-4943A; KW-4679
Trademarks: Opatanol (Alcon); Patanol (Alcon)
Molecular Formula: C21H23NO3.HCl
Molecular Weight: 373.87
Percent Composition: C 67.46%, H 6.47%, N 3.75%, O 12.84%, Cl 9.48%
Properties: Crystals from acetone-water, mp 248° (dec). Sol in water.
Melting point: mp 248° (dec)
////////////
Paper
Journal of the Brazilian Chemical Society
J. Braz. Chem. Soc. vol.25 no.12 São Paulo Dec. 2014
http://dx.doi.org/10.5935/0103-5053.20140255
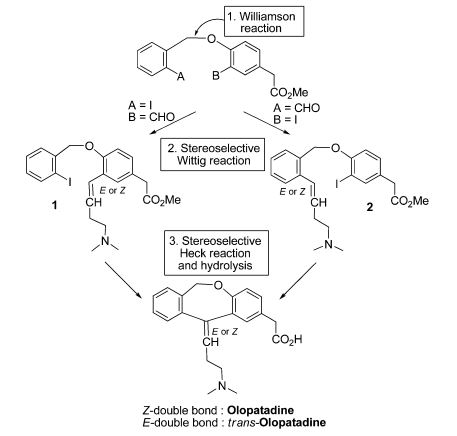
An intramolecular Heck-based cyclization was used as a key step for commendable synthesis of the antihistaminic drug olopatadine (133) and its trans isomer (134).67 Besides the Heck reaction, another vital step in this route was a stereoselective Wittig olefination using a non-stabilized phosphorus ylide that afforded the olefins 135 and 136 (E:Z ratio = 9:1 for 135, for instance). Concerning the Heck reaction, Pd(OAc)2, K2CO3, and NBu4Cl (TBAC) were allowed to react with 135 and 136 at 60 ºC during 24 h, providing the cyclic adducts 137 and 138 with reasonable 60% and 55% yields, respectively. However, it is important to note that in catalytic terms, the results were not encouraging, considering that 20 mol% of palladium was used and a disappointing turnover number (TON) of 3 was observed (Scheme 37).
In relation to the stereochemistry of the Heck products, the above results were not surprising since they were consistent with a syn-insertion of the arylpalladium intermediate (provided by oxidative addition step) at the olefinic moiety followed by a syn β-elimination that afforded the product with the ascribed stereochemistry. Finally, with the cyclic products in hands, the syntheses were completed by alkaline hydrolysis of methyl esters that afforded the target olapatadine and trans-olapatadine.
67 Bosch, J.; Bachs, J.; Gómez, A. M.; Griera, R.; Écija, M.; Amat, M.; J. Org. Chem.2012, 77 , 6340.
In relation to the stereochemistry of the Heck products, the above results were not surprising since they were consistent with a syn-insertion of the arylpalladium intermediate (provided by oxidative addition step) at the olefinic moiety followed by a syn β-elimination that afforded the product with the ascribed stereochemistry. Finally, with the cyclic products in hands, the syntheses were completed by alkaline hydrolysis of methyl esters that afforded the target olapatadine and trans-olapatadine.

67 Bosch, J.; Bachs, J.; Gómez, A. M.; Griera, R.; Écija, M.; Amat, M.; J. Org. Chem.2012, 77 , 6340.
The construction of a new bond between sp2- and sp-hybridized carbons is known as the Sonogashira reaction,48and it is nowadays a widely employed methodology for the construction of arylacetylenes.3,49,50 For example, a Sonogashira coupling was employed by the research and development group of Kyowa Hakko Kirin in a new and concise synthetic route for olopatadine hydrochloride (92), a commercial anti-allergic drug that was previously developed by the same company.51
The reported synthesis goes through the Sonogashira reaction between the easy accessible aryl halide 93 and alkyne 94 leading to adduct 95 in 94% yield. This adduct is then subjected to a second metal-catalyzed transformation, a stereospecific palladium-catalyzed intramolecular cyclization, whose optimum conditions were identified based on an elegant and comprehensive Design of Experiments (DoE) investigation to provide 96(Scheme 28).

Elaboration of the cyclization product 96 through aminomethylation and ester hydrolysis followed by acid work-up completes the synthesis of the final target. Although the presented synthetic route is very promising and concise, providing olopatadine hydrochloride in 54% overall yield for 6 steps from commercially available materials, it has so far been reported only on a laboratory scale (5 g for the Sonogashira coupling and 200 mg for the cyclization step).
51 Nishimura, K.; Kinugawa, M.; Org. Process Res. Dev.2012, 16 , 225