ARKIVOC Volume 2006
Part (v): Commemorative Issue in Honor of
Facilitator: Luba Ignatovich
Scientific Editor: Mikael Begtrup
Part (v): Commemorative Issue in Honor of
Facilitator: Luba Ignatovich
Scientific Editor: Mikael Begtrup
2. The structure of Omeprazole in the solid state: a 13C and 15N NMR/CPMAS study (EL-1719AP)
Rosa M. Claramunt, Concepción López and José Elguero
Full Text: PDF (193K)
pp. 5 - 11
Rosa M. Claramunt, Concepción López and José Elguero
Full Text: PDF (193K)
pp. 5 - 11
The structure of Omeprazole in the solid state: a 13C and 15N NMR/CPMAS study
Rosa M. Claramunt,a Concepción López,a and José Elguero b *
a Departamento de Química Orgánica y Bio-Orgánica, Facultad de Ciencias, UNED, Senda del Rey 9, E-28040 Madrid, Spain
b Instituto de Química Médica, CSIC, Juan de la Cierva, 3. E-28006 Madrid, Spain E-mail: iqmbe17@iqm.csic.es
To our friend Professor Edmunds Lukevics on his 70th anniversary
 Edmunds Lukevics
Edmunds Lukevics
Abstract
The 13C and 15N CPMAS spectra of a solid sample of Omeprazole have been recorded and all the signals assigned. The sample consists uniquely of the 6-methoxy tautomer. For analytical purposes, the signals of the other tautomer, the 5-methoxy one, were estimated from the data in solution (Magn. Reson. Chem. 2004, 42, 712).
Keywords: Omeprazole, NMR, 13C, 15N, CPMAS, tautomerism, benzimidazole
see at
Edmunds LUKEVICS
(14.12.1936 - 21.11.2009)
 | Professor Edmunds LUKEVICS Latvian Institute of Organic Synthesis, Head of the Laboratory of Organometallic Chemistry
Aizkraukles iela 21,
Riga, LV-1006 Latvia |
Born: December 14, 1936, Liepaja, Latvia
Departed: November 21, 2009, Riga, Latvia
Departed: November 21, 2009, Riga, Latvia
Interests:
- Organometallic Compounds
- Heterocyclic Compounds
- Biological Activity of Organic Compounds
Main Research:
Development of methods for the synthesis of organosilicon and -germanium derivatives of furan, thiophene and nitrogen-containing heterocycles ; study of the influence of organosilicon ,-germanium and -tin substituents on the direction of substitution and addition reactions of furan and thiophene derivatives ; study of hydrosilylation and hydrogermylation reactions, synthesis and investigation of properties of penta- and hexacoordinated organosilicon and -germanium derivatives; application of alkenyl silanes and germanes in the synthesis of nitrogen-containing heterocycles; application of phase-transfer catalysis and ultrasonic irradiation in organometallic synthesis; synthesis of biologically active organosilicon and organogermanium compounds and studies of their properties.
Education:
- University of Latvia (Faculty of Chemistry), 1958
- Dr.chem. (Candidate of Science in former USSR, Ph.D. in Western countries), Latvian Academy of Sciences, Riga, 1966
- Dr.habil.chem. (Doctor of Science in former USSR), Latvian Academy of Sciences, Riga, 1973
Experience:
Latvian Institute of Organic Synthesis -
- Junior Researcher, 1958-1967
- Senior Researcher, 1968-1970
- Head, Laboratory of Organometallic Chemistry, 1970 - 2009
- Vice-director, 1980-1982
- Director, 1982 - 2003
Honours and Awards:
- Corresponding Member, Latvian Academy of Sciences , 1982
- Full Member, Latvian Academy of Sciences , 1987
- Member, New York Academy of Sciences, 1993
- The Latvian Academy of Sciences Gustavs Vanags Prize (in Chemistry), 1986
- Latvian SSR State Prize, 1974, 1989
- S.Hiller Medal (Latvian Institute of Organic Synthesis), 1990
- G.Vanags Medal (Riga Technical University), 1991
- D.H.Grindel Medal (company ‘Grindex’, Latvia), 1995
- L.Liepina Medal (Institute of Inorganic Chemistry, Riga), 1996
- The Latvian Academy of Sciences Grand Medal, 1996
- Silver Medal of Milan University, 1996
- Schmiedebergs Medal (Latvian Pharmacological Society), 1998
- The Latvian Academy of Sciences and Company "GRINDEX" Prize, 1999
- Paul Walden's Medal (Riga Technical University), 2000
- Latvian Academy of Sciences Presidium Award, 1971, 1973, 1977, 1981, 1982, 1985,1987, 1989, 1992
- International Man of the Year (The International Biographic Centre of Cambridge, England), 1992-1993, 1994-1995
- Man of the Year (The American Biographical Institute), 1994, 2005
- The first-level Badge of Honour of the Order of Three Stars, 1997
- Company “Grindex” gold badge of honour, 2001
- The Cabinet of Ministers of the Republic of Latvia Prize , 2004
- American Medal of Honor (ABI), 2005
- Gold Medal for Latvia (ABI), 2006
- The Plato Award (IBC), 2006
- Man of Achievement (ABI), 2007
Professional Activities:
- Member of Presidium and Senate, Latvian Academy of Sciences, 1987-1991
- Member of Board, Division of Chemical and Biological Sciences, Latvian Academy of Sciences, 1983-1993
- Member, Latvian Academy of Sciences Commission on Terminology, 1987- 1999
- Chairman, Habilitation and Promotion Council (Chemistry and Pharmacy), Latvian Institute of Organic Synthesis, 1994 -1999
- Member (Chairman,1991-1993, 1997-2002), Latvian Council of Science Expert Committee for Chemistry, 1991 - 2006
- Vice-chairman, Habilitation and Promotion Council (Chemistry), University of Latvia, 1998- 2009
- Member of Editorial Board for:
Khimiya Geterotsikicheskikh. Soedinenii (Chemistry of Heterocyclic Compounds, Springer), 1980-1985; Editor-in-chief, 1985 - 2009
Proceedings of Latvian Academy of Sciences, 1982-1990
Latvian Journal of Chemistry, 1991 - 2009
Bioorganicheskaya Khimiya, 1989 - 1993
Applied Organometallic Chemistry, 1990 - 2009
Main Group Metal Chemistry, 1992 - 2009
Metal-Based Drugs, 1993 - 2003
Mendeleev Communications, 1994 - 2009
Advances in Heterocyclic Chemistry, 1994 - 2009
Silicon Chemistry, 2001-2007
Arkivoc, 2001 - 2009
Bioinorganic Chemistry and Applications, 2003 - 2006
Heterocyclic Communications, 2005 - 2009
Molecules, 2008 - 2009
Journal of Organic and Pharmaceutical Chemistry (Ukraine), 2009 - 2009
Proceedings of Latvian Academy of Sciences, 1982-1990
Latvian Journal of Chemistry, 1991 - 2009
Bioorganicheskaya Khimiya, 1989 - 1993
Applied Organometallic Chemistry, 1990 - 2009
Main Group Metal Chemistry, 1992 - 2009
Metal-Based Drugs, 1993 - 2003
Mendeleev Communications, 1994 - 2009
Advances in Heterocyclic Chemistry, 1994 - 2009
Silicon Chemistry, 2001-2007
Arkivoc, 2001 - 2009
Bioinorganic Chemistry and Applications, 2003 - 2006
Heterocyclic Communications, 2005 - 2009
Molecules, 2008 - 2009
Journal of Organic and Pharmaceutical Chemistry (Ukraine), 2009 - 2009
- Chairman, Scientific Council “Chemistry and Technology of Sulfur Organic Compounds”, USSR State Committee of Science and Technics, 1982-1987
- Chairman, Council “Application of Organometallic Compounds in National Economy”, USSR (Russian) Academy of Sciences, 1984-1992
- Member, United Libraries Informative Council, USSR Academy of Sciences, 1985-1990
- Member, Scientific Council “Physiologically Active Compounds”, USSR Academy of Sciences, 1986-1992
- Member, Scientific and Technical Council, USSR Ministry of Medical and Microbiological Industry, 1987-1990
- Member, Soviet National Committee on collecting and estimating information in science and technics “CODATA”, 1987-1990
- Member of Council for Coordination of scientific work, Department of Biochemistry, Biophysics and Physiologically Active Compounds, USSR Academy of Sciences, 1988-1991
- Member of International Organizing Committees
- International Conference on the Coordination and Organometallic Chemistry of Germanium, Tin and Lead, 1992, 1995, 1998, 2001
- International Symposium on Organosilicon Chemistry, 1993, 1996, 1999, 2002, 2005, 2008.
Memberships:
- Member of Organometallic Chemistry Division, Federation of European Chemical Societies, 1995-2005
- Member of Organometallic Chemistry Division, European Association for Chemical & Molecular Sciences, 2006
- Member, Latvian Chemical Society, 1995
- Member, American Chemical Society, 1997
- Member, National Geographic Society, 1997
- Honorary Member, Pharmacological Society of Latvia, 1998
Lectures
Invited Lectures at Universities
- Indian Institute of Science, Bangalore (India), 1989
- Indian Institute of Technology, Bombay (India), 1989
- University of Dresden (Germany), 1989
- Universities of Bordeaux, Tolouse, Montpellier, Marseilles (France), 1990, 1994
- University of Lund (Sweden), 1992
- University of Alcala de Henares ( Spain), 1993
- Tohoku University (Sendai, Japan), 1991, 1992
- Tokyo University of Science (Japan), 1997
- Kyoto University (Japan), 1997
- Universities of Kyoto and Kanagawa, Japan, 2002.
Invited Lectures and Symposium’s Plenary Lectures:
- 40th Nobel Symposium (Lidingö, Sweden), 1977
- VI Symposium on Chemistry of Heterocyclic Compounds (Brno, Czechoslovakia), 1978
- 7th International Symposium on Organosilicon Chemistry (Kyoto, Japan), 1984
- VI FECHEM Conference on Organometallic Chemistry (riga, Latvia), 1985
- II Soviet-Indian Symposium on Organometallic Chemistry( Irkutsk, Russia), 1989
- 17th DDR-Poland Colloquy on Organometallic Chemistry (Holzhau, Germany), 1989
- 6th International Conference on Organometallic and Coordination Chemistry of Germanium, Tin and Lead (Brussels, Belgium), 1989
- Huang Minlon Symposium on Organic Chemistry (Shanghai, China), 1989
- International Chemical Conference on Silicon and Tin ( Kuala Lumpur, Malaisia), 1989
- 9th International Symposium on Organosilicon Chemistry (Edinburgh, UK), 1990
- 1st Meeting of the European Society of Sonochemistry, Autrans (Grenoble, France), 1990
- 11th International Symposium on Medicinal Chemistry (Jerusalem, Israel), 1990
- S.Hiller Memorial Lectures (Riga, Latvia), 1990
- 1st Meeting of Japanese Germanium Discussion Group (Tokyo, Japan), 1991
- International Conference on Environmental and Biological Aspects of Maingroups Organometals (Padua, Italy), 1991
- 3rd Swedish-German workshop: Nucleic Acid Synthesis, Structure and Function (Uppsala, Sweden), 1992
- 2nd ANAIC Conference on Materials Science and Environmental Chemistry of Main Group Elements (Kual Lumpur, Malaysia), 1993
- Todai Symposium “Ge-Sn-Pb Tokyo’93”: International Symposium on Organic, Bioorganic and Bioinorganic Chemistry of Compounds of higher row Group 14-elements (Tokyo, Japan), 1993
- 10th International Symposium on Organosilicon Chemistry (Poznan, Poland), 1993
- 3rd Meeting of the European Society of Sonochemistry (Figueira da Foz, Portugal), 1993
- 14th Nordic Meeting of Structural Chemists (Helsinki, Finland), 1993
- 8th International Conference on the Organometallic Chemistry of Germanium, Tin and Lead (Sendai, Japan), 1995
- 8th IUPAC Symposium on Organometallic Chemistry Directed Towards Organic Synthesis (Santa Barbara, USA), 1995
- 8th Symposium Heterocycles in Bioorganic Chemistry (Como, Italy), 1996
- 9th International Conference on the Coordination and Organometallic Chemistry of Germanium, Tin, and Lead (Melbourne, Australia), 1998
- 12th International Conference on Organosilicon Chemistry (Sendai, Japan), 1999
- International Conference on Organic Synthesis "Balticum Organicum Sinteticum-2000"(Vilnius, Lithuania), 2000
- X International Symposium "Jubilee Krka Prizes" (Novo Mesto, Slovenia), 2000
Recent/Representative Publications:
- E.Ya. Lukevits, M.G.Voronkov. Organic Insertion Reactions of Group IV Elements, 1966, New York: Consultants Bureau, 413 pp.
- S.N.Borisov, M.G.Voronkov, E.Ya.Lukevits. Organosilicon Heteropolymers and Heterocompounds, 1970, NewYork: Plenum Press, 633 pp.
- S.N.Borisov, M.G.Voronkov, E.Ya.Lukevits. Organosilicon Derivatives of Phosphorus and Sulfur, 1971, NewYork; London: Plenum Press, 343 pp.
- M.G.Voronkov, G.I..Zelchan, E.Ya.Lukevits. Silizium und Leben, 1975, Berlin: Akademie-Verlag, 370 pp.
- E.Lukevics, O.Pudova, R.Sturkovich. Molecular Structure of Organosilicon Compounds, 1989, Chichester: Ellis Horwood Ltd., 359 pp.
- E.Lukevics, T.Gar, L.Ignatovich, V.Mironov. Biological Activity of Germanium Compounds, 1990, Riga: Zinatne, 191 pp. (in Russian).
- E.Lukevics, A.Zablocka. Nucleoside Synthesis: Organosilicon Methods, 1991, Chichester: Ellis Horwood, 496 pp.
- E.Lukevics, L.Ignatovich. Biological activity of organogermanium compounds. - In: The Chemistry of Organic Germanium, Tin and Lead Compounds/Ed. Z.Rappoport/, Wiley, Chichester, 2002, vol. 2, pt. 2, pp. 1653-1683.
- E.Lukevics, O.Pudova. Biological activity of organogermanium compounds. - In: The Chemistry of Organic Germanium, Tin and Lead Compounds/Ed. Z.Rappoport/, Wiley, Chichester, 2002, vol. 2, pt. 2, pp. 1685-1714.
- E.Lukevics, O.Pudova. Silyl imidic esters. - In: Science of Synthesis, Thieme, 2002, vol. 4, pp. 305-315.
- E. Lukevics, P. Arsenyan, S. Belyakov, O. Pudova. Synthesis, structure and chemical transformations of ethynylgermatranes – Eur. J. Inorg. Chem., 2003, Iss.17, pp.3139-3143.
- R. Abele, E. Abele, M. Fleisher, S. Grinberga, E. Lukevics. Novel fluoride ion mediated synthesis of unsymmetrical siloxanes under phase transfer catalysis conditions. – J. Organomet. Chem., 2003, vol.686, N 1/2, pp.52-57.
- E. Lukevics, L. Ignatovich, I.Shestakova. Synthesis, psychotropic and anticancer activity of 2,2-dimethyl-5-[5-trialkylgermyl(silyl)-2’-hetarylidene]-1,3-dioxane-4,6-diones and their analogues. - Appl.Organomet. Chem., 2003, vol. 17, N 12, pp.898-905.
- P. Arsenyan, K. Rubina, I. Shestakova, E. Abele, R. Abele, I. Domracheva, A. Nesterova, J. Popelis, E. Lukevics. Synthesis and cytotoxicity of silylalkylthio-substituted N-heterocycles and their hydroselenites. – Appl. Organomet. Chem., 2003, vol. 17, N 11, pp.825-830.
- E. Lukevics, L. Ignatovich, T. Shul’ga, S. Belyakov. The crystal structure of 2-benzo[b]thienylgermatrane. – Appl. Organomet. Chem., 2003, vol. 17, N 9, pp.745-746.
- K. Rubina, E. Abele, P. Arsenyan, M. Fleisher, J. Popelis, A. Gaukhman, E. Lukevics. The role of palladium catalyst and base in stereoselective tranformations of (E)-2-chlorovinylsulfides. –Tetrahedron, 2003, vol.59, N 38, pp.7603-7607.
- I. Iovel, L. Golomba, J. Popelis, S. Grinberga, E. Lukevics Catalytic hydrosilylation of furan, thiophene, and pyridine aldimines. – Chem. Heterocycl. Comp., 2003, vol.39, N 1, pp.49-55.
- G. Veinberg, M. Vorona, I. Shestakova, I. Kanepe, E. Lukevics. Design of ß-lactams with mechanism based nonbacterial activities. – Current Medicinal Chemistry, 2003, vol.10, N 17, pp.1741-1757.
- E. Lukevics, P. Arsenyan, O. Pudova. Methods for the synthesis of oligothiophenes. – Heterocycles, 2003, vol.60, N 3, pp.663-687.
- V.Dirnens, V.Klusa, J.Skuyins, S.Svirskis, S.Germane, A.Kemme, E.Lukevics. Synthesis and pharmacological activity of silyl isoxazolines-2. - Silicon Chemistry, 2003 (publ. 2004), vol. 2, N 1/2, pp. 11-25.
- I.Iovel, L.Golomba, M.Fleischer, J.Popelis, S.Grinberga, E.Lukevics. Hydrosilylation of (hetero)aromatic aldimines in the presence of Pd(I) complex. - Chem.Heterocycl. Comp., 2004, vol. 40, N 6, pp. 701-714.
- P.Arsenyan, O.Pudova, J.Popelis, E.Lukevics. Novel radial oligothienylsilanes. - Tetrahedron Lett., 2004, vol. 45, N 15, pp. 3109-3111.
- E.Lukevics, L.Ignatovich, S.Belyakov. Crystallographic report: 2-furfurylgermatrane. - Appl. Organomet. Chem., 2004, vol. 18, N 4, p. 203.
- G. Veinberg, I. Shestakova, M. Vorona, I. Kanepe, E. Lukevics. Synthesis of antitumor 6-alkylidenepenicillanate sulfones and related 3-alkylidene-2-azetidinones. - Bioorg. Med. Chem. Letters, 2004, vol. 14, No 1, 147-150.
- E.Lukevics, L.Ignatovich, T.Shulga, S.Belyakov. 1-[4-(2-Thienyl)phenyl]germatrane. - Appl. Organomet. Chem., 2005, vol. 19, N 1, pp. 167-168.
- E.Lukevics, L.Ignatovich. Biological activity of organosilicon compounds. - In: Metallotherapeutic Drugs and Metal-Based Diagnostic Agents. The Use of Metals in Medicine / Eds. M.Gielen, E.R.T.Tiekink/, 2005, J.Wiley & Sons, Ltd. Chichester, pp. 83-107.
- E.Lukevics, L.Ignatovich. Biological activity of organogermanium compounds. - In: Metallotherapeutic Drugs and Metal-Based Diagnostic Agents. The Use of Metals in Medicine / Eds. M.Gielen, E.R.T.Tiekink/, 2005, J.Wiley & Sons, Ltd. Chichester, pp. 279-295.
- Yu.Melnik, M.Vorona, G.Veinberg, J.Popelis, L.Ignatovich, E.Lukevics. Synthesis and stereoisomerization of 2-(1-alkoxyimino-2,2,2-trifluoroethyl)-5-trimethylsilylfurans. - Chem. Heterocycl. Comp., 2005, vol. 41, N 6, pp. 718-721.
- L.Ignatovich, J.Popelis, E.Lukevics. Synthesis and NMR spectra of diaryl- and dihetarylsilacycloalkanes. - In: Organosilicon Chemistry VI / Eds. N.Auner and J.Weis/, Wiley-VCH Weinheim, 2005, vol. 1, pp. 559-562.
- L.Ignatovich, D.Zarina, I.Shestakova, S.Germane, E.Lukevics. Synthesis and bological activity of silicon derivatives of 2-trifluoroacetylfuran and their oximes. - In: Organosilicon Chemistry VI / Eds. N.Auner and J.Weis/, Wiley-VCH Weinheim, 2005, vol. 1, pp. 563-568.
- E.Lukevics, L.Ignatovich, I.Sleiksha, I.Shestakova, I.Domrachova, J.Popelis. Synthesis and cytotoxic activity of silacycloalkylsubstituted heterocyclic aldehydes. - Appl. Organomet. Chem., 2005, vol. 19, N 10, pp. 1109-1113.
- S.Belyakov, E.Alksnis, V.Muravenko, I.Turovskis, J.Popelis, E.Lukevics. Crystal structure and conformation of 8-(2-hydroxyethylamino)- and 8-(pyrrolidin-1-yl)adenosines. - Nucleosides, Nucleotides & Nucleic Acid, 2005, vol. 24, N 8, pp. 1199-1208.
- A. Zablotskaya, I.Segal, S.Belyakov, E.Lukevics. Silyl modification of biologically active compounds. 11. Synthesis, physico-chemical and biological evaluation of N-(trialkoxysilylalkyl)tetrahydro(iso,silaiso)quinoline derivatives. Appl. Organomet. Chem. 2006, vol.20, N 2, 149-159.
- A.Zablotskaya, I.Segal, J.Popelis, E.Lukevics, S.Baluja, I.Shestakova, I.Domracheva. Silyl modification of biologically active compounds. 12. Silyl group as true incentive to antitumour and antibacterial action of choline and colamine analogues. - Appl. Organomet. Chem. 2006, vol. 20, N 11, 721-728.
- E.Lukevics, L.Ignatovich, I.Sleiksha, V.Muravenko, I.Shestakova, S.Belyakov, J.Popelis. Synthesis, structure and cytotoxic activity of 2-acetyl-5-trimethylsilylthiophene(furan) and their oximes. - Appl. Organomet. Chem. 2006, vol 20, N 7, 454-458.
- L.Ignatovich, V.Muravenko, S.Grinberga, E.Lukevics. Novel reactions to form an Si-O-Ge group. - Chem.Heterocycl. Comp., 2006, vol. 42, N 2, 268-271.
- E.Lukevics, I.Shestakova, I.Domrachova, A.Nesterova, Y.Ashaks, D.Zaruma. Synthesis of complex compounds of methyl derivatives of 8-quinolineselenol with metals and their cytotoxic activity. - Chem.Heterocycl. Comp., 2006, vol. 42, N 1, 53-59.
- E.Lukevics, L.Ignatovich, I.Sleiksha, V.Romanov, S.Grinberga, J.Popelis, I.Shestakova. A New method for the synthesis of silicon- and germanium-containing 2-acetylfurans and 2-acetylthiophenes. -Chem.Heterocycl. Comp., 2007, vol. 43, N 2, 143-150.
- V.Dirnens, I.Skrastina, J.Popelis, E.Lukevics. Synthesis of isoxazolinylxanthines. - Chem.Heterocycl. Comp., 2007, vol. 43, N 2, 193-196.
- E.Lukevics, L.Ignatovich, S.Belyakov. Disordering in the crystal structure of thienylgermatranes. - Chem.Heterocycl. Comp., 2007, vol. 43, N 2, 243-249.
- E.Lukevics, I.Shestakova, I.Domrachova, E. Yashchenko, D.Zaruma. Y.Ashaks. Cytotoxic di(8-quinolyl)disulfides. - Chem.Heterocycl. Comp., 2007, vol. 43, N 5, 629-633.
- V.M.Vorona, I.Potorocina, G.Veinberg, I.Shestakova, I.Kanepe, M.Petrova, E. Liepinsh, E.Lukevics. Synthesis and structural modification of tert-butyl ester of 7a-chloro-2-(N,N-dimethylaminomethylene)-3-methyl-1,1-dioxoceph-3-em carboxylic acid.- Chem.Heterocycl. Comp., 2007, vol. 43, N 5, 646-652.
- A.Zablotskaya, I.Segal, E.Lukevics, S.Belyakov, H.Spies. Tetrahydroquinoline and tetrahydroisoquinoline mixed ligand rhenium complexes with the SNS/S donor atom set.- Appl.Organomet.Chem.,2007, vol.21, N 4, 288-293.
- A.Zablotskaya, I.Segal, M. Maiorov, D. Zablotsky, A. Mishnev E.Lukevics, I.Shestakova, I. Domracheva. Synthesis and characterization of nanoparticles with an iron oxide magnetic core and a biologically active trialkylsilylated aliphatic alkanolamine shell. J. Magn. Magn. Mater. 2007, 311, pp. 135-139.
- Zablotskaya A., Segal I., Lukevics E., Maiorov M., Zablotsky D., Blums E., Shestakova I., Domracheva I. Synyhesis, physico-chemical and biological study of trialkylsiloxyalkylamine coated iron oxide/oleic acid magnetic nanoparticles for the treatment of cancer. - Appl. Organomet. Chem. 2008, vol. 22, pp. 82-88.
- E.Lukevics, E.Abele. Four-membered rings with three heteroatoms not including oxygen, sulfur or nitrogen atom. - In: Comprehensive Heterocyclic Chemistry III., 2008, 2. Four-membered heterocycles together with all fused systems containing a four-membered heterocyclic ring (Exec. Ed. A. Katritzky, FRS: Eds Ch.A. Ramsden, E.V.Scriven, R.J.Taylor), pp. 973-989.
- Soualami S., Ignatovich L., Lukevics E.,Ourari A., Jouikov V. Electrochemical oxidation of benzylgermatranes. - J. Organomet. Chem., 2008, vol.693 (7), pp. 1346-1352.
- Lukevics E., Ignatovich L., Shul'ga T., Belyakov S. Synthesis and crystal structure of 1-(4-fluorophenyl)- and 1-(4-dimethylamino)phenylgermatranes. - Chem. Heterocycl.Comp. (Engl.Ed.), 2008, vol. 44 (5), pp. 615-620.
- Abele E., Lukevics E. Synthesis of Heterocycles from Oximes. - In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. (Eds. Z.Rapoport, J.F.Liebmann ), J.Wiley, Chichester, 2009, Part I, pp. 233-302.
- Erchak N., Belyakov S., Kalvinsh I., Pypowski K., Valbahs E., Lukevics E. Two polymorphic modifications of 1-(N-morpholiniomethyl)spirobi(3-oxo-2,5-dioxa-1-silacyclopentan)ate hydrate. -Chem.Heterocycl. Comp.(Engl. Ed.), 2009, vol. 45, N 9, pp.1137-1143..
- Zablotskaya A.,Segal I., Lukevics E., Maiorov M., Zablotsky D., Blums E., Shestakova I., Domracheva I. Water-soluble magnetic nanoparticleswith biologically active stabilizers. - J.Magn.Mater.,2009, 321, pp. 1428-1432.
- Ignatovich L., Muravenko V., Shestakova I., Domracheva I, Popelis J., Lukevics E. Synthesis and Cytotoxic activity of new 2-[(3-aminopropyl)- dimethylsilyl]-5-triethylsilylfurans. - Appl. Organomet. Chem. 2009, DOI 10.1002, aoc, 1538.
- Vorona M., Veinberg G.,Liepinsh E., Kazoka H., Andrejeva G., Lukevics E. Enzymatic synthesis of amoxycilloic acids. - Chem.Heterocycl. Comp.(Engl. Ed.), 2009, vol. 45, N 6, pp.782-754.
- Zablotskaya A.,Segal I., Lukevics E. Iron oxide-based magnetic nanostructures bearing cytotoxic organosilicon molecules for drug delivery and therapy. - Appl. Organomet. Chem. 2010, vol. 24, N 3, pp. 150-157.
- Ignatovich L., Muravenko V., Shestakova I., Domracheva I, Popelis J., Lukevics E. Synthesis and Cytotoxic activity of new 2-[(3-aminopropyl)- dimethylsilyl]-5-triethylsilylfurans. - Appl. Organomet. Chem. 2010, vol. 24, N 3, pp. 158-161.
- Segal I., Zablotskaya A.,Lukevics E., Maiorov M., Zablotsky D., Blums E., Mishnew A., Georgieva R., Shestakova I., Gulbe A. Preparation and cytotoxic properties of goethite-based nanoparticles covered with decyldimethyl(dimethylaminoethoxy)silane metoxyde. - Appl. Organomet. Chem. 2010, vol. 24, N 3, pp. 193-197.
- Ignatovich L., Muravenko V., RomanovsV, Sleiksha I., Shestakova I., Domracheva I, Belyakov S., Popelis J., Lukevics E. Synthesis, structure and cytotoxic activity of new 1-[5-organylsilyl(germyl)-2-furyl(thienyl)]nitroethenes. - Appl. Organomet. Chem. 2010, vol. 24, N 12, pp. 858-864.
- Lukevics E., Abele E., Ignatovich L. Biologically Active Silacyclanes. - Adv. Heterocycl. Chem., 2010, vol. 99, pp. 107-141.
- Abele E., Lukevics E. Synthesis, structure and reactions of organometallic derivatives of oximes. - In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Eds. by Zvi Rapoport, J.F.Liebman. 2011, Vol.2, Part 1 (Chapter 4), pp. 145-203.
- Katkevics M., Kukosha T., Lukevics E. Heterocycles from hydroxylamines and hydroxamic acids. - In: The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids. Eds. by Zvi Rapoport, J.F.Liebman. 2011, Vol.2, Part 1 (Chapter 5), pp. 205-293.
Research Projects:
- E.Lukevics (Head of Project). Silylheterocycles in Organic Chemistry. Latvian Council of Science (1993-1995).
- E.Lukevics (Head of Project). Bifunctional Organosilicon Compounds. Latvian Council of Science (1993-1995).
- E.Lukevics (Head of Project). Synthesis of Heterocyclic Organosilicon and Organogermanium Compounds, Investigation of their Physical and Chemical Properties. Latvian Council of Science (1997-2000 ).
- E.Lukevics (Head of Project). Asymmetric and Catalytic Synthesis of Heteroaromatic Compounds. Latvian Council of Science (1997-2000 ).
- E.Lukevics (Head of Program). The Development of Modern Methods of Organic Chemistry Directed towards the Development of Pharmaceutical Industry in Latvia. Latvian Council of Science (1997-2000 ).
- E.Lukevics (Head of Project). Experimental and Theoretical Aspects of the Catalytical Synthesis of Heteroaromatic Compounds. Latvian Council of Science (2001 -2004 ).
- E.Lukevics (Head of Project). Comparative Study of the Structure and Biological Activity of Organosilicon and Organogermanium Compounds. Latvian Council of Science (2001 - 2004).
- E.Lukevics (Head of Project). Heterocyclic Derivatives of Tetra- and Hypercoordinated Germanium and Silicon. Latvian Council of Science (2005 -).
- E.Lukevics ( Programme Director). Development of Organic Synthesis Methods for Obtaining of Biologically Active Compounds. Latvian Council of Science (2002 -2005 ).
- E.Lukevics ( Programme Director). Development of Heteroatom Chemistry for Preparation of Biologically Active Compounds. Latvian Council of Science (2006 - 2009 ).
- E.Lukevics (Head of Project). Carbofunctional Silylheterocycles. Latvian Council of Science (2009 ).
Hobbies:
Opera, Basketball, Mountains.
Edmunds LUKEVICS | Edmunds LUKEVICS Head of Laboratory of Organometallic Chemistry
Latvian Institute of Organic Synthesis
Born: December 14, 1936, Liepaja, Latvia
Departed: November 21, 2009, Riga, Latvia |
Interests in inventing:
Main invention:In the sphere of medicament synthesis:
In the sphere of the synthesis of agricultural chemicals:
Selection of patent documents:
Totally: 104 authors' certificates of USSR, 11 patents of Latvia, 3 patents of Germany, 3 patents of Canada, 3 patents of France, 3 patents of Italy, 1 patent of Japan, 1 patent of Switzerland, 4 patents of Great Britain, 5 patents of USA.
Patents of Latvia:
| |
Riga latvia


The building of the Brotherhood of Blackheads is one of the most iconic buildings of Old Riga (Vecrīga)

RIGA

RIGA

RIGA

RIGA

Cook in traditional latvian dress serving local food for tourists Riga Latvia





")
")
")
")
")
 COCK WILL TEACH YOU
COCK WILL TEACH YOU





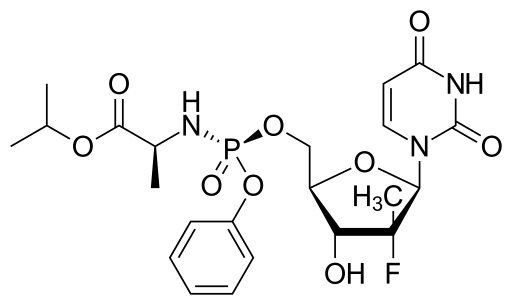
![Propan-2-yl (2s)-2-[[[(2r,3r,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_tlE-1tWVSUM0T3MUNorrjKwxZjl-z1Cmd8f8T_Wtg0Ba1gxpbMTcTqAqZhRz1m1sLERHb51xpu_11e1cK5_6kz777djtJQ6XCk0mGOzlF-9BP-9ZTk0_a8LvV6Fg=s0-d)
![propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate NMR spectra analysis, Chemical CAS NO. 1190307-88-0 NMR spectral analysis, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate H-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_tVAl2pFZuY92NksNEyv-d4IsESyg-hve-kvRhtb76aIBTgLrWmRureKyjXFQ_Ef3D6liP8ys4iGRnsYGLAn65c-rHQGEoVaNue81UAE2HyDpntePqO1Qec3e6hFWmU1LxNUCascEGj8txqcQ=s0-d)
![propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate NMR spectra analysis, Chemical CAS NO. 1190307-88-0 NMR spectral analysis, propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate C-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_uyqQ1Sw5xdsTi30QgsYIQ_hHGvpqqTneVfHikI2YzBcIjoA9yVdeMs5M3YmFzaKi75wR9Pgi4jjmvfI8_sxuCNx7Fdl-xnYVym9pkVpYQrKDlZVe6tb3Ie4T-de9o6CNjxt--cOYJ1hF7ho80=s0-d)