Lisdexamfetamine
- Molecular FormulaC15H25N3O
- Average mass263.379 Da
608137-32-2 [RN]
Elvanse [Trade name]
Hexanamide, 2,6-dia
Lisdexamfetamine
L-lysine-d-amphetam
(2S)-2,6-diamino-N-
H645GUL8KJ
L-lysine-(+)-amphet
NRP104|Vyvanse®
UNII:H645GUL8KJ
UNII-H645GUL8KJ
Vyvanse®

(2S)-2,6-Diamino-N-[(1S)-1-methyl-2-phenylethyl]hexanamide dimethanesulfonate
| Applicants: | NEW RIVER PHARMACEUTICALS INC. [US/US]; The Governor Tyler, 1881 Grove Avenue, Radford, VA 24141 (US) (For All Designated States Except US). MICKLE, Travis [US/US]; (US) (For US Only). KRISHNAN, Suma [US/US]; (US) (For US Only). MONCRIEF, James, Scott [US/US]; (US) (For US Only). LAUDERBACK, Christopher [US/US]; (US) (For US Only). MILLER, Christal [US/US]; (US) (For US Only) |
| Inventors: | MICKLE, Travis; (US). KRISHNAN, Suma; (US). MONCRIEF, James, Scott; (US). LAUDERBACK, Christopher; (US). MILLER, Christal; (US) |

MICKLE, Travis
Dr. Travis Mickle founded KemPharm, Inc. in late 2006. Prior to KemPharm, from January 2003 to October 2005, Dr. Mickle was Director of Drug Discovery and Chemical Development at New River Pharmaceuticals where he also served in a variety of other senior research roles since joining the firm in 2001. During his tenure at New River, Dr. Mickle was responsible for creating a strong preclinical and clinical pipeline of drugs in the areas of ADHD, pain and thyroid dysfunction. His contributions included, as principal inventor, the discovery and development of lisdexamfetamine dimesylate, the highly successful therapy for the treatment of ADHD known as Vyvanse®. In addition, Dr. Mickle was an active participant in FDA and DEA meetings representing the company’s discovery/chemistry group and was also called in as a critical scientific resource during New River’s financings and strategic partnering discussions. Before his departure, Dr. Mickle played an integral part in New River’s development into a successful publicly-traded company which was subsequently acquired for $2.6 billion by its marketing partner, Shire PLC. Dr. Mickle is also the author of more than 150 US and international patents and patent applications, as well as several research papers. Dr. Mickle holds a Ph.D in Bio-Organic Chemistry from the University of Iowa.
Dr. Travis Mickle founded KemPharm, Inc. in late 2006. Prior to KemPharm, from January 2003 to October 2005, Dr. Mickle was Director of Drug Discovery and Chemical Development at New River Pharmaceuticals where he also served in a variety of other senior research roles since joining the firm in 2001. During his tenure at New River, Dr. Mickle was responsible for creating a strong preclinical and clinical pipeline of drugs in the areas of ADHD, pain and thyroid dysfunction. His contributions included, as principal inventor, the discovery and development of lisdexamfetamine dimesylate, the highly successful therapy for the treatment of ADHD known as Vyvanse®. In addition, Dr. Mickle was an active participant in FDA and DEA meetings representing the company’s discovery/chemistry group and was also called in as a critical scientific resource during New River’s financings and strategic partnering discussions. Before his departure, Dr. Mickle played an integral part in New River’s development into a successful publicly-traded company which was subsequently acquired for $2.6 billion by its marketing partner, Shire PLC. Dr. Mickle is also the author of more than 150 US and international patents and patent applications, as well as several research papers. Dr. Mickle holds a Ph.D in Bio-Organic Chemistry from the University of Iowa.
ABOUT SUMA KRISHNAN
Mrs. Krishnan has served as our Senior Vice President — Regulatory Affairs since 2012. From 2009 to 2011, Mrs. Krishnan served as Senior Vice President of Product Development at Pinnacle Pharmaceuticals, Inc. From 2007 to 2009, she served as Chief Financial Officer of Light Matters Foundation. Previously, Mrs. Krishnan was Vice President, Product Development at New River Pharmaceuticals Inc. from September 2002 until its acquisition by Shire plc in April 2007.
Mrs. Krishnan has 22 years’ experience in drug development. Prior to serving at New River Pharmaceuticals Inc., Mrs. Krishnan served in the following capacities: Director, Regulatory Affairs at Shire Pharmaceuticals, Inc., a specialty pharmaceutical company; Senior Project Manager at Pfizer, Inc., a multi-national pharmaceutical company; and a consultant at the Weinberg Group, a pharmaceutical and environmental consulting firm.
Mrs. Krishnan began her career as a discovery scientist for Janssen Pharmaceuticals, Inc., a subsidiary of Johnson & Johnson, a multi-national pharmaceutical company, in May 1991. Mrs. Krishnan received an M.S. in Organic Chemistry from Villanova University, an M.B.A. from Institute of Management and Research (India) and an undergraduate degree in Organic Chemistry from Ferguson University (India).
Director, Regulatory Affairs
Lisdexamfetamine (contracted from L-lysine-dextroamphetamine) is a prodrug of the central nervous system (CNS) stimulantdextroamphetamine, a phenethylamine of the amphetamine class that is used in the treatment of attention deficit hyperactivity disorder (ADHD) and binge eating disorder.[4][5] Its chemical structure consists of dextroamphetamine coupled with the essential amino acid L-lysine. Lisdexamfetamine itself is inactive prior to its absorption and the subsequent rate-limited enzymaticcleavage of the molecule's L-lysine portion, which produces the active metabolite (dextroamphetamine).
Lisdexamfetamine can be prescribed for the treatment of attention deficit hyperactivity disorder (ADHD) in adults and children six and older, as well as for moderate to severe binge eating disorder in adults.[4] The safety and the efficacy of lisdexamfetamine dimesylate in children with ADHD three to five years old have not been established.[6]
Lisdexamfetamine is a Class B/Schedule II substance in the United Kingdom and a Schedule II controlled substance in the United States (DEA number 1205)[7] and the aggregate production quota for 2016 in the United States is 29,750 kilograms of anhydrous acid or base.[8] Lisdexamfetamine is currently in Phase III trials in Japan for ADHD.[9]
Uses
Medical
Lisdexamfetamine is used primarily as a treatment for attention deficit hyperactivity disorder (ADHD) and binge eating disorder;[4] it has similar off-label uses as those of other pharmaceutical amphetamines.[4][5] Long-term amphetamine exposure at sufficiently high doses in some animal species is known to produce abnormal dopamine system development or nerve damage,[10][11] but, in humans with ADHD, pharmaceutical amphetamines appear to improve brain development and nerve growth.[12][13][14] Reviews of magnetic resonance imaging (MRI) studies suggest that long-term treatment with amphetamine decreases abnormalities in brain structure and function found in subjects with ADHD, and improves function in several parts of the brain, such as the right caudate nucleus of the basal ganglia.[12][13][14]
Reviews of clinical stimulant research have established the safety and effectiveness of long-term continuous amphetamine use for the treatment of ADHD.[15][16][17] Randomized controlled trials of continuous stimulant therapy for the treatment of ADHD spanning two years have demonstrated treatment effectiveness and safety.[15][17] Two reviews have indicated that long-term continuous stimulant therapy for ADHD is effective for reducing the core symptoms of ADHD (i.e., hyperactivity, inattention, and impulsivity), enhancing quality of life and academic achievement, and producing improvements in a large number of functional outcomes[note 1] across nine outcome categories related to academics, antisocial behavior, driving, non-medicinal drug use, obesity, occupation, self-esteem, service use (i.e., academic, occupational, health, financial, and legal services), and social function.[16][17] One review highlighted a nine-month randomized controlled trial in children with ADHD that found an average increase of 4.5 IQ points, continued increases in attention, and continued decreases in disruptive behaviors and hyperactivity.[15] Another review indicated that, based upon the longest follow-up studies conducted to date, lifetime stimulant therapy that begins during childhood is continuously effective for controlling ADHD symptoms and reduces the risk of developing a substance use disorder as an adult.[17]
Current models of ADHD suggest that it is associated with functional impairments in some of the brain's neurotransmitter systems;[18] these functional impairments involve impaired dopamine neurotransmission in the mesocorticolimbic projection and norepinephrine neurotransmission in the noradrenergic projections from the locus coeruleus to the prefrontal cortex.[18]Psychostimulants like methylphenidate and amphetamine are effective in treating ADHD because they increase neurotransmitter activity in these systems.[19][18][20] Approximately 80% of those who use these stimulants see improvements in ADHD symptoms.[21] Children with ADHD who use stimulant medications generally have better relationships with peers and family members, perform better in school, are less distractible and impulsive, and have longer attention spans.[22][23] The Cochrane Collaboration's reviews[note 2] on the treatment of ADHD in children, adolescents, and adults with pharmaceutical amphetamines stated that while these drugs improve short-term symptoms, they have higher discontinuation rates than non-stimulant medications due to their adverse side effects.[25][26] A Cochrane Collaboration review on the treatment of ADHD in children with tic disorders such as Tourette syndrome indicated that stimulants in general do not make tics worse, but high doses of dextroamphetamine could exacerbate tics in some individuals.[27]
Individuals over the age of 65 were not commonly tested in clinical trials of lisdexamfetamine for ADHD.[4] Lisdexamfetamine is being investigated for possible treatment of cognitive impairment associated with schizophrenia and excessive daytime sleepiness.[28]
Cognitive
In 2015, a systematic review and a meta-analysis of high quality clinical trials found that, when used at low (therapeutic) doses, amphetamine produces modest yet unambiguous improvements in cognition, including working memory, long-term episodic memory, inhibitory control, and some aspects of attention, in normal healthy adults;[29][30] these cognition-enhancing effects of amphetamine are known to be partially mediated through the indirect activation of both dopamine receptor D1 and adrenoceptor α2 in the prefrontal cortex.[19][29] A systematic review from 2014 found that low doses of amphetamine also improve memory consolidation, in turn leading to improved recall of information.[31] Therapeutic doses of amphetamine also enhance cortical network efficiency, an effect which mediates improvements in working memory in all individuals.[19][32]Amphetamine and other ADHD stimulants also improve task saliency (motivation to perform a task) and increase arousal (wakefulness), in turn promoting goal-directed behavior.[19][33][34] Stimulants such as amphetamine can improve performance on difficult and boring tasks and are used by some students as a study and test-taking aid.[19][34][35]Based upon studies of self-reported illicit stimulant use, 5–35% of college students use diverted ADHD stimulants, which are primarily used for performance enhancement rather than as recreational drugs.[36][37][38] However, high amphetamine doses that are above the therapeutic range can interfere with working memory and other aspects of cognitive control.[19][34]
Physical
Amphetamine is used by some athletes for its psychological and athletic performance-enhancing effects, such as increased endurance and alertness;[39][40] however, non-medical amphetamine use is prohibited at sporting events that are regulated by collegiate, national, and international anti-doping agencies.[41][42] In healthy people at oral therapeutic doses, amphetamine has been shown to increase muscle strength, acceleration, athletic performance in anaerobic conditions, and endurance (i.e., it delays the onset of fatigue), while improving reaction time.[39][43][44] Amphetamine improves endurance and reaction time primarily through reuptake inhibition and effluxion of dopamine in the central nervous system.[43][44][45] Amphetamine and other dopaminergic drugs also increase power output at fixed levels of perceived exertion by overriding a "safety switch" that allows the core temperature limit to increase in order to access a reserve capacity that is normally off-limits.[44][46][47] At therapeutic doses, the adverse effects of amphetamine do not impede athletic performance;[39][43] however, at much higher doses, amphetamine can induce effects that severely impair performance, such as rapid muscle breakdown and elevated body temperature.[48][49][43]
Available forms
Vyvanse capsules are available in doses of 10 mg, 20 mg, 30 mg, 40 mg, 50 mg, 60 mg, and 70 mg of the active ingredient, lisdexamfetamine dimesylate.[50] Vyvanse capsules contain several inactive ingredients, including microcrystalline cellulose, croscarmellose sodium, and magnesium stearate.[50] The capsule shells contain gelatin and titanium dioxide, and may contain FD&C Red 3, FD&C Yellow 6, FD&C Blue 1, black iron oxide, and yellow iron oxide.[50]













DESCRIPTION/SYNTHESIS
Lisdexamfetamine dimesylate is approved and marketed in the United States for the treatment of attention-deficit hyperactivity disorder in pediatric patients. The active compound lisdexamfetamine contains D-amphetamine covalently linked to the essential amino acid L-lysine. Controlled release of D-amphetamine, a psychostimulant, occurs following administration of lisdexamfetamine to a patient. The controlled release has been reported to occur through hydrolysis of the amide bond linking D-amphetamine and L-lysine.
A procedure for making lisdexamfetamine hydrochloride is described in U.S. Pat. No. 7,223,735 to Mickle et al. (hereinafter Mickle). The procedure involves reacting D-amphetamine with (S)-2,5-dioxopyrrolidin-1-yl 2,6-bis(tert-butoxycarbonylamino)hexanoate to form a lysine-amphetamine intermediate bearing tert-butylcarbamate protecting groups. This intermediate is treated with hydrochloric acid to remove the tert-butylcarbamate protecting groups and provide lisdexamfetamine as its hydrochloride salt. However, this procedure suffers several drawbacks that are problematic when carrying out large scale reactions, such as manufacturing scale, to prepare lisdexamfetamine.




Lisdexamphetamine of formula I, is a conjugate of D-amphetamine and L-lysine and is chemically named as (2S)-2,6-diamino-N-[(lS)-methyl-2-phenylethyl]hexan amide.
Formula- I

Amphetamines stimulate central nervous system (CNS). Amphetamine is prescribed for treatment of various disorders, including attention deficit hyperactivity disorder (ADHD), obesity, nacrolepsy. It is approved as lisdextamphetamine dimesylate of formula IA and
Formula- IA

marketed under trade name Vyvanse for treatment of attention-deficit hyperactivity disorder in pediatric patients.
L-Lysine-D-amphetamine and its pharmaceutically acceptable salts were first disclosed in US patent 7,662,787 wherein it is exemplified as hydrochloride salt. Process for preparation of L- lysine-D-amphetamine includes reaction of BOC-Lys-(BOC)-hydroxysuccinimido ester with D-amphetamine in dioxane using diisopropyl ethyl amine (DIPEA) as a base to obtain BOC- protected lisdexamphetamine which is then purified using flash chromatography and further reacted with a mixture of 4M hydrochloric acid /dioxane to yield L-lysine-D-amphetamine hydrochloride. The process is as shown in following scheme:

4M HCI/dioxane

In equivalent patent US 7,659,253, process for preparation of mesylate salt is disclosed as shown below.

NHS;DCC;
isopropyl acetate
-50°C, 2 hr

Process includes preparation of BOC-Lys-(BOC)-hydroxysuccinimido ester wherein use of reagents like N-hydroxy-succinimide (NHS) and Ν,Ν-dicyclohexyl- carbodimiide (DCC) is carried out, The above processes involve use of flash column chromatography to purify crude BOC- protected L-lysine-D-amphetamine intermediate. Use of column chromatography is very cumbersome, tedoius and time consuming, therefore not advisable at commercial scale. Further Ν,Ν-dicyclohexylcarbodimiide is known to be highly toxic and moisture sensitive compound, and its use leads to formation of a large amount of Ν,Ν-dicyclohexyl urea (DCU) as bye product which has to be removed from reaction mixture.. Therefore use of DCC is not advisable at industrial scale.
International patent publication WO 2010/042120 discloses a process for preparing L-lysine- D-amphetamine or its salts by reacting D-amphetamine with protected lysine or its salt by using an alkylphosphonic acid anhydride as coupling agent in presence of a base and solvent. The process is as shown in following scheme:

The application discloses use of alkylphosphonic acid anhydrides, which are expensive, and needs additional testing to show absence of phosphic impurities in intermediate or final compound to meet regulatory requirements. So it is not appealing to use alkylphosphonic anhydrides for scale up operations.
International patent publication WO2010/148305 discloses a process for preparation of lisdexamphetamine by removal of chlorine from Ν,Ν'-bistrifluoroacetyl-chloro- lisdexamphetamine by using hydrogenation catalyst like Pd/C, under hydrogen gas to form Ν,Ν'-bistrifluoroacetyl-lisdexamphetamine which on further deprotection by using deprotecting agent to form lisdexamphetamine. Alternatively first deprotection by using deprotecting agent and then chlorine is removed by using hydrogenation catalyst like Pd/C under hydrogen gas. The process involves additional steps of inserting chloro group and thereafter removing chloro group; further Pd/C is an expensive reagent, hence not attractive option from cost point of view.
It is therefore, necessary to overcome problems associated with prior art and to provide an efficient process for preparation of lisdexamphetamine and its pharmaceutically acceptable salts using easily available, less expensive, easy to handle raw materials and avoid use of column chromatography.
PATENT





- Example IPreparation of N,N′-Biscarbobenzyloxy-Lisdexamfetamine (LDX-(Cbz)2)

General Experimental Procedure: In an appropriately sized, inert jacketed reactor charge 378.3 g of Cbz-Lys(Cbz)-OSu and 2309 g of isopropyl acetate. Heat the stirred slurry to ˜50° C. In a second reactor mix 100.0 g of D-amphetamine into 165 g of isopropyl acetate. Add the D-amphetamine solution to the batch over 1.75-2.25 hours. After the addition is complete stir the heterogeneous mixture at 50-55° C. until the reaction is complete by HPLC analysis. Charge 1197 g of methanol and heat the batch at a vigorous reflux 1 hour. Cool the batch to 45-55° C. over 2 hours then hold at temperature for 14-16 hours. Cool the batch to 15-25° C. at a rate of 5-10° C. per hour. Filter the slurry. Rinse the wet cake with 449 g of methanol and dry on the filter under nitrogen. To a clean, dry reactor charge the crude solids and 1642 g of methanol. Stir the slurry and heat to a vigorous reflux for 2 hours. Cool the batch to 45-55° C. over 1-2 hours then hold at temperature 12-16 hours. Continue cooling to 15-25° C. at a rate of 5-10° C. per hour. Filter the slurry and wash the wet cake with 547 g of methanol. Vacuum dry the wet cake at ˜55° C. to give product as a white to off-white solid (332 g, 88 mol %).
The crude LDX-(Cbz)product isolated from the reaction mixture by crystallization had a purity of 99.96% according to HPLC analysis.1H NMR analysis of crude LDX-(Cbz)2product revealed the presence of 0.57% by weight N-hydroxysuccinimide. The purified LDX-(Cbz)2product obtained by re-crystallization from methanol had a purity of 99.99% according to HPLC analysis.1H NMR analysis of the purified LDX-(Cbz)2product obtained by re-crystallization from methanol revealed that the amount of N-hydroxysuccinimide in the purified LDX-(Cbz)2product was reduced to 0.05% by weight.
Example 2Preparation of Lisdexamfetamine Dimesylate

General Experimental Procedure: In an appropriately sized, inert autoclave charge 100.0 g of LDX-(Cbz)2, 1 g of 10% (50% wet) palladium on carbon and 607.5 g of n-butanol. Stir the mixture under 100-150 psi of hydrogen at 80-85° C. until the reaction is complete by HPLC analysis. Heat the batch to 95-97° C. and hot filter. Transfer the product rich filtrate to an appropriately sized glass reactor. Charge 7.6 g of methanesulfonic acid maintaining a batch temperature of 32-38° C. To the resulting solution add 1.3 g of LDX-2MSA seed crystal. Stir the batch 4-16 hours at 32-38° C. Charge 30.4 g of methanesulfonic acid to the slurry over not less than 2 hours maintaining a batch temperature of 32-38° C. After the addition stir 1-2 hours at 32-38° C. Charge 436 g of isopropyl acetate over not less than 2 hours, then stir 1-2 hours at 32-38° C. Cool the batch to 15-25° C. and hold 1 hour. Filter the slurry. Wash the wet cake with a premixed combination of n-butanol (91.5 g) and isopropyl acetate (32.7 g) followed by a wash of isopropyl acetate (87.2 g). Vacuum dry the wet cake at ˜50° C. to give product as a white to off-white solid (78.8 g, 92 mol %).
PATENT
Sun Pharmaceutical Industries Ltd
Process for preparation of lisdexamphetamine and its salts via a novel aziridine intermediate is claimed. Lisdexamphetamine attention deficit hyperactivity disorder (ADHD). Represents the first patenting to be seen from Sun pharmaceutical that focuses on lisdexamphetamine. At the time of publication Pawar and Patel are affiliated with Chattem chemicals .


PATENT
IN 2011DE02040
WO 2013011526
IN 2009CH01986
WO 2010042120

Formula II A
Formula IV
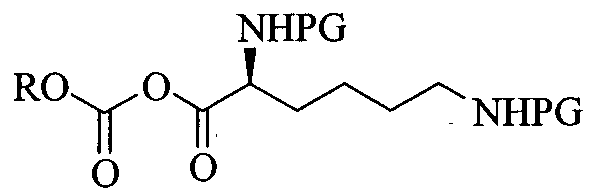
Formula IVA
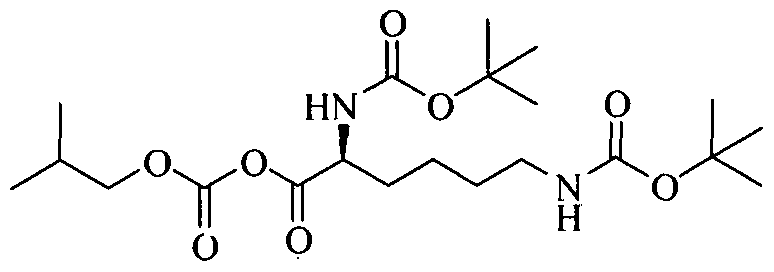
EXAMPLES
Examplel Preparation of 2,6-bis-tertiarvbutoxycarbonylamino hexanoic acid
To a solution of L-lysine monohydrochloride (25g, 0.14mol) and sodium hydroxide (15g) in water (250 ml), ditertiary butyl dicarbonate (70.0 g, 0.32 mol) was added at 15-25°C. The temperature was slowly raised to 55-60 °C and the reaction mixture was stirred for 12 hours.. After completion of reaction, ( monitored by TLC), the reaction mixture was cooled to 10-
15°C and pH was adjusted to 2.5-3.5 with 2N hydrochloric acid. The reaction mass' was then extracted with dichloromethane (2 x 125 ml) and combined organic layer was successively washed with water (150 ml) and brine (150 ml). Dichloromethane layer was distilled under vacuum at 30-40 °C to obtain 29.7g of title compound as a viscous oily mass having purity 96.5% by HPLC.
Example 2: Preparation of (5-tert-butoxycarbonylamino-5-(l-methyl-2-phenyl- ethylcarba moyl)-pentyll-carbamic acid tert-butyl ester;
To a solution of 2,6-bis-tertbutoxy carbonylamino hexanoic acid (7.5g) in dichloromethane (150 ml) , triethyl amine (8.0 ml) was added at 25-30°C and the reaction mixture was stirred for 15 minutes. The solution was cooled to -15 to -10°C and isobutylchloroformate (4.35 g) was slowly added under nitrogen atmosphere and stirred for 30 minutes at -15°C to - 10°C. A solution of D-amphetamine (3.85 g) in dichloromethane (10 ml) was slowly added and. the reaction mixture was stirred at 15 to -10°C for 60 minutes. The reaction completion was checked by TLC. After completion of the reaction temperature was raised to 25-30°C and reaction mixture was successively washed with 0.5 N hydrochloric acid solution (2 x 75 ml), sodium bicarbonate solution (5%w/w 75ml), water (50 ml) and brine solution (50 ml). The combined dichloromethane layer was dried over sodium sulfate (10.0 g) and distilled at 30- 40°C to obtain a semisolid compound which was stirred with a mixture of n-heptane (85 ml) and ethyl acetate (5ml) at 25-30°C for 30 minutes. The solid, thus obtained, was filtered and dried to get 10.21 g of title compound having purity 89.77% by HPLC. The crude compound was dissolved in ethanol (45ml) at 50-55°C and water (50ml) was added. The reaction mixture was slowly cooled to 35-40°C, stirred for 30 minutes. The solid, thus obtained, was filtered and dried to get 7.35g of pure title compound as a white crystalline solid having purity 99.5 % by HPLC.
Example 3: Preparation of lisdexamphetamine dimesylate
(5-Tert-butoxycarbonylamino-5-( 1 -methyl-2-phenyl-ethylcarbamoyl)-pentyl]-carbamic acid tert-butyl ester (2.5g,) was dissolved in a mixture of isopropyl alcohol (10ml) and ethyl acetate (10ml) at 40-45°C and the reaction mass was cooled to 15-20°C. To this cold solution, methane sulphonic acid (2.5g) was added slowly and stirred for 12 hours at 15- 20°C. The reaction completion was checked by HPLC. The resulting solid was filtered, washed with a mixture of chilled isopropyl alcohol (5ml) and ethyl acetate (5ml) and dried under vacuum to obtain 1.68 g of title compound as a white crystalline solid having purity 99.72 % by HPLC.
Example 4: Preparation of lisdexamphetamine dimesylate:
(5-Tert-butoxycarbonylamino-5-( 1 -methyl-2-phenyl-ethylcarbamoyl)-pentyl]-carbamic acid tert-butyl ester (2.5 g,) was dissolved in ethanol (20 ml) at 25-30°C. To the reaction mixture, methane sulphonic acid (2.5 g) was slowly added and the reaction mixture was heated to 55- 60°C and stirred for 3 hours at 55-60°C. The reaction mixture was cooled to 25-30°C, stirred for 2 hours, filtered, washed with ethanol (10ml) and dried under vacuum to obtain 1.55g of lisdexamphetamine dimesylate having purity 99.61 % by HPLC.
Example 5: Purification of lisdexamphetamine dimesylate
Lisdexamphetamine dimesylate (1.40g,) was dissolved in ethanol (10 ml) at 50-55 °C and ethyl acetate (10 ml) was slowly added at 50-55 °C. The reaction mixture was cooled to 20- 25°C and stirred for 30 minutes. The resulting solid was filtered, washed with a mixture of ethanol and ethyl acetate (3 ml, 1: 1) and dried under vacuum at 55-60°C to obtain 1.28g of pure lisdexamphetamine dimesylate as a white crystalline solid having purity 99.90 % by HPLC.
Example 6: Preparation of lisdexamphetamine dimesylate
To a stirred solution of L-lysine monohydrochloride (50g) and sodium hydroxide (30 g) in water (500ml) at 15-25°C, ditertbutyl dicarbonate(140g) was added. The temperature was slowly raised to 55-60°C and reaction mixture was stirred for 12 hours. After completion of reaction, the reaction mixture was cooled to 10-15°C and pH was adjusted to 2.5-3.5 with 2N hydrochloric acid. The reaction mixture was then extracted with dichloromethane (2 x 250 ml) and combined dichloromethane layer was successively washed with water (300 ml) and brine (300 ml). To the organic layer triethyl amine (58g) in dichloromethane (500 ml) was added. The solution was cooled to -15 to -20°C and isobutylchloroformate (42.5 g) was slowly added at -15 to -20°C and stirred for 1 hour. A solution of D-amphetamine (41.85 g) in dichloromethane (100 ml) was slowly added to reaction mixture at -15 to -20 °C and stirred. After completion of the reaction, the reaction mixture was successively washed with 0.5 N hydrochloric solution (2 x 450 ml), sodium bicarbonate solution (5%w/w, 450 ml), water (450 ml) and brine solution (450 ml). The organic layer was dried over sodium sulfate and distilled under vacuum at 30-40°C to afford a residue. To this residue, ethanol (480 ml) was added followed by slow addition of methane sulphonic acid (55 g) under nitrogen atmosphere. The reaction temperature was raised 55-60°C and after completion of reaction, the reaction mixture was cooled to 20-25°C and stirred for 2 hours. The resulting solid was filtered, washed with ethanol (50 ml) and suck dried for 30 minutes, further washed with ethanol and dried to obtain lisdexamphetamine dimesylate.
Mechanism of action
Pharmacodynamics of amphetamine in a dopamine neuron

via AADC
Amphetamine enters the presynaptic neuron across the neuronal membrane or through DAT. Once inside, it binds to TAAR1 or enters synaptic vesicles through VMAT2. When amphetamine enters synaptic vesicles through VMAT2, it collapses the vesicular pH gradient, which in turn causes dopamine to be released into the cytosol (light tan-colored area) through VMAT2. When amphetamine binds to TAAR1, it reduces the firing rate of the dopamine neuron via potassium channels and activates protein kinase A (PKA) and protein kinase C (PKC), which subsequently phosphorylate DAT. PKA-phosphorylation causes DAT to withdraw into the presynaptic neuron (internalize) and cease transport. PKC-phosphorylated DAT may either operate in reverse or, like PKA-phosphorylated DAT, internalize and cease transport. Amphetamine is also known to increase intracellular calcium, an effect which is associated with DAT phosphorylation through a CAMKIIα-dependent pathway, in turn producing dopamine efflux.
Lisdexamfetamine is an inactive prodrug that is converted in the body to dextroamphetamine, a pharmacologically active compound which is responsible for the drug’s activity.[119] After oral ingestion, lisdexamfetamine is broken down by enzymes in red blood cells to form L-lysine, a naturally occurring essential amino acid, and dextroamphetamine.[4] The conversion of lisdexamfetamine to dextroamphetamine is not affected by gastrointestinal pH and is unlikely to be affected by alterations in normal gastrointestinal transit times.[4][120]
The optical isomers of amphetamine, i.e., dextroamphetamine and levoamphetamine, are TAAR1 agonists and vesicular monoamine transporter 2 inhibitors that can enter monoamine neurons;[121][122] this allows them to release monoamine neurotransmitters (dopamine, norepinephrine, and serotonin, among others) from their storage sites in the presynaptic neuron, as well as prevent the reuptake of these neurotransmitters from the synaptic cleft.[121][122]
Lisdexamfetamine was developed with the goal of providing a long duration of effect that is consistent throughout the day, with reduced potential for abuse. The attachment of the amino acid lysine slows down the relative amount of dextroamphetamine available to the blood stream. Because no free dextroamphetamine is present in lisdexamfetamine capsules, dextroamphetamine does not become available through mechanical manipulation, such as crushing or simple extraction. A relatively sophisticated biochemical process is needed to produce dextroamphetamine from lisdexamfetamine.[120] As opposed to Adderall, which contains roughly equal parts of racemic amphetamine and dextroamphetamine salts, lisdexamfetamine is a single-enantiomer dextroamphetamine formula.[119][123] Studies conducted show that lisdexamfetamine dimesylate may have less abuse potential than dextroamphetamine and an abuse profile similar to diethylpropion at dosages that are FDA-approved for treatment of ADHD, but still has a high abuse potential when this dosage is exceeded by over 100%.[120]
Pharmacokinetics
The oral bioavailability of amphetamine varies with gastrointestinal pH;[118] it is well absorbed from the gut, and bioavailability is typically over 75% for dextroamphetamine.[124] Amphetamine is a weak base with a pKa of 9.9;[125]consequently, when the pH is basic, more of the drug is in its lipid soluble free base form, and more is absorbed through the lipid-rich cell membranes of the gut epithelium.[125][118] Conversely, an acidic pH means the drug is predominantly in a water-soluble cationic (salt) form, and less is absorbed.[125] Approximately 15–40% of amphetamine circulating in the bloodstream is bound to plasma proteins.[126]
The half-life of amphetamine enantiomers differ and vary with urine pH.[125] At normal urine pH, the half-lives of dextroamphetamine and levoamphetamine are 9–11 hours and 11–14 hours, respectively.[125] An acidic diet will reduce the enantiomer half-lives to 8–11 hours; an alkaline diet will increase the range to 16–31 hours.[127][128] The biological half-life is longer and distribution volumes are larger in amphetamine dependent individuals.[128] The immediate-release and extended release variants of salts of both isomers reach peak plasma concentrations at 3 hours and 7 hours post-dose respectively.[125] Amphetamine is eliminated via the kidneys, with 30–40% of the drug being excreted unchanged at normal urinary pH.[125] When the urinary pH is basic, amphetamine is in its free base form, so less is excreted.[125] When urine pH is abnormal, the urinary recovery of amphetamine may range from a low of 1% to a high of 75%, depending mostly upon whether urine is too basic or acidic, respectively.[125] Amphetamine is usually eliminated within two days of the last oral dose.[127]
The prodrug lisdexamfetamine is not as sensitive to pH as amphetamine when being absorbed in the gastrointestinal tract;[129] following absorption into the blood stream, it is converted by red blood cell-associated enzymes to dextroamphetamine via hydrolysis.[129] The elimination half-life of lisdexamfetamine is generally less than one hour.[129]
CYP2D6, dopamine β-hydroxylase (DBH), flavin-containing monooxygenase 3 (FMO3), butyrate-CoA ligase (XM-ligase), and glycine N-acyltransferase (GLYAT) are the enzymes known to metabolize amphetamine or its metabolites in humans.[sources 9] Amphetamine has a variety of excreted metabolic products, including 4-hydroxyamphetamine, 4-hydroxynorephedrine, 4-hydroxyphenylacetone, benzoic acid, hippuric acid, norephedrine, and phenylacetone.[125][127][134] Among these metabolites, the active sympathomimetics are 4‑hydroxyamphetamine,[138] 4‑hydroxynorephedrine,[139] and norephedrine.[140] The main metabolic pathways involve aromatic para-hydroxylation, aliphatic alpha- and beta-hydroxylation, N-oxidation, N-dealkylation, and deamination.[125][127] The known metabolic pathways, detectable metabolites, and metabolizing enzymes in humans include the following:
Metabolic pathways of amphetamine in humans[sources 9]

Para-
Hydroxylation
Hydroxylation
Para-
Hydroxylation
Hydroxylation
Para-
Hydroxylation
Hydroxylation
unidentified
Beta-
Hydroxylation
Hydroxylation
Beta-
Hydroxylation
Hydroxylation
Oxidative
Deamination
Deamination
Oxidation
unidentified
Glycine
Conjugation
Conjugation
The primary active metabolites of amphetamine are 4-hydroxyamphetamine and norephedrine;[134] at normal urine pH, about 30–40% of amphetamine is excreted unchanged and roughly 50% is excreted as the inactive metabolites (bottom row).[125] The remaining 10–20% is excreted as the active metabolites.[125] Benzoic acid is metabolized by XM-ligase into an intermediate product, benzoyl-CoA,[136] which is then metabolized by GLYAT into hippuric acid.[137]
Chemistry
Comparison to other formulationsLisdexamfetamine dimesylate is a water-soluble (792 mg/mL) powder with a white to off-white color.[50]
Lisdexamfetamine dimesylate is one marketed formulation delivering dextroamphetamine. The following table compares the drug to other amphetamine pharmaceuticals.
| drug | formula | molecular mass [note 8] | amphetamine base [note 9] | amphetamine base in equal doses | doses with equal base content [note 10] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (g/mol) | (percent) | (30 mg dose) | ||||||||
| total | base | total | dextro- | levo- | dextro- | levo- | ||||
| dextroamphetamine sulfate[142][143] | (C9H13N)2•H2SO4 | |||||||||
| amphetamine sulfate[144] | (C9H13N)2•H2SO4 | |||||||||
| Adderall | ||||||||||
| 25% | dextroamphetamine sulfate[142][143] | (C9H13N)2•H2SO4 | ||||||||
| 25% | amphetamine sulfate[144] | (C9H13N)2•H2SO4 | ||||||||
| 25% | dextroamphetamine saccharate[145] | (C9H13N)2•C6H10O8 | ||||||||
| 25% | amphetamine aspartate monohydrate[146] | (C9H13N)•C4H7NO4•H2O | ||||||||
| lisdexamfetamine dimesylate[147] | C15H25N3O•(CH4O3S)2 | |||||||||
| amphetamine base suspension[note 11] | C9H13N | |||||||||
History, society, and culture
Lisdexamfetamine was developed by New River Pharmaceuticals, who were bought by Shire Pharmaceuticals shortly before lisdexamfetamine began being marketed. It was developed for the intention of creating a longer-lasting and less-easily abused version of dextroamphetamine, as the requirement of conversion into dextroamphetamine via enzymes in the red blood cells increases its duration of action, regardless of the route of ingestion.[148] The drug lisdexamfetamine dimesylate is the first prodrug of its kind.
On 23 April 2008, Vyvanse received FDA approval for the adult population.[149] On 19 February 2009, Health Canada approved 30 mg and 50 mg capsules of lisdexamfetamine for treatment of ADHD.[150] On 8 February 2012, Vyvanse received FDA approval for maintenance treatment of adult ADHD.[151] In February 2014, Shire announced that two late-stage clinical trials had shown that Vyvanse was not an effective treatment for depression.[152] Lisdexamfetamine was granted approval in a number of European countries for the treatment of ADHD in children and adolescents over the age of 6 years, as well as adults who are continuing treatment from childhood, after a positive outcome of the regulatory procedure.[153] Shire also recently announced receipt of a positive result from a European decentralised procedure for lisdexamfetamine for adult patients with ADHD in the United Kingdom, Sweden and Denmark, expanding the indication of lisdexamfetamine to include newly diagnosed adult patients.[154]
In January 2015, lisdexamfetamine was approved by the U.S. Food and Drug Administration for treatment of binge eating disorder in adults.[28][155][156]
In January 2017, a new dosage form of lisdexamfetamine in the form of a chewable tablet (as opposed to a capsule) was approved by the FDA.[157]
Brand names
Clinical research
Some clinical trials that used lisdexamfetamine as an add-on therapy with a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI) for treatment-resistant depression indicated that this is no more effective than the use of an SSRI or SNRI alone.[159] Other studies indicated that psychostimulants potentiated antidepressants, and were under-prescribed for treatment resistant depression. In those studies patients showed significant improvement in energy, mood, and psychomotor activity.[160]
Notes
- Jump up^ The ADHD-related outcome domains with the greatest proportion of significantly improved outcomes from long-term continuous stimulant therapy include academics (~55% of academic outcomes improved), driving (100% of driving outcomes improved), non-medical drug use (47% of addiction-related outcomes improved), obesity (~65% of obesity-related outcomes improved), self esteem (50% of self-esteem outcomes improved), and social function (67% of social function outcomes improved).[16]The largest effect sizes for outcome improvements from long-term stimulant therapy occur in the domains involving academics (e.g., grade point average, achievement test scores, length of education, and education level), self-esteem (e.g., self-esteem questionnaire assessments, number of suicide attempts, and suicide rates), and social function (e.g., peer nomination scores, social skills, and quality of peer, family, and romantic relationships).[16]Long-term combination therapy for ADHD (i.e., treatment with both a stimulant and behavioral therapy) produces even larger effect sizes for outcome improvements and improves a larger proportion of outcomes across each domain compared to long-term stimulant therapy alone.[16]
- Jump up^ Cochrane Collaboration reviews are high quality meta-analytic systematic reviews of randomized controlled trials.[24]
- Jump up^ The 95% confidence interval indicates that there is a 95% probability that the true number of deaths lies between 3,425 and 4,145.
- Jump up^ Transcription factors are proteins that increase or decrease the expression of specific genes.[93]
- Jump up^ In simpler terms, this necessary and sufficient relationship means that ΔFosB overexpression in the nucleus accumbens and addiction-related behavioral and neural adaptations always occur together and never occur alone.
- Jump up^ NMDA receptors are voltage-dependent ligand-gated ion channels that requires simultaneous binding of glutamate and a co-agonist (d-serine or glycine) to open the ion channel.[105]
- Jump up^ The review indicated that magnesium L-aspartate and magnesium chloride produce significant changes in addictive behavior;[71] other forms of magnesium were not mentioned.
- Jump up^ For uniformity, molecular masses were calculated using the Lenntech Molecular Weight Calculator[141] and were within 0.01g/mol of published pharmaceutical values.
- Jump up^ Amphetamine base percentage = molecular massbase / molecular masstotal. Amphetamine base percentage for Adderall = sum of component percentages / 4.
- Jump up^ dose = (1 / amphetamine base percentage) × scaling factor = (molecular masstotal / molecular massbase) × scaling factor. The values in this column were scaled to a 30 mg dose of dextroamphetamine sulfate. Due to pharmacological differences between these medications (e.g., differences in the release, absorption, conversion, concentration, differing effects of enantiomers, half-life, etc.), the listed values should not be considered equipotent doses.
- Jump up^ This product (Dyanavel XR) is an oral suspension (i.e., a drug that is suspended in a liquid and taken by mouth) that contains 2.5 mg/mL of amphetamine base. The amphetamine base contains dextro- to levo-amphetamine in a ratio of 3.2:1, which is approximately the ratio in Adderall. The product uses an ion exchange resin to achieve extended release of the amphetamine base.
PATENT CITATIONS
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US20050038121 * | Jun 1, 2004 | Feb 17, 2005 | New River Pharmaceuticals Inc. | Abuse resistant lysine amphetamine compounds |
| US20110196173 * | Oct 9, 2008 | Aug 11, 2011 | Andreas Meudt | Process for the Synthesis of Amphetamine Derivatives |
| DE1493824A1 * | Nov 23, 1964 | May 22, 1969 | Hoffmann La Roche | Verfahren zur Herstellung von Aminocarbonsaeureamiden |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | "Benzyl Chloroformate" in Handbook of Reagents for Organic Synthesis - Activating Agents and Protecting Groups ; Pearson et al., eds., 1999 John Wiley & Sons, pp. 46-50 |
| 2 | * | DATABASE CAPLUS CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; Database Accession No. 1966:19805, Abstract NL 6414901, 28 July 1965 |
| 3 | * | Smith and March. Advanced Organic Chemistry 6th ed. (501-502) |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8614346 | Jun 18, 2010 | Dec 24, 2013 | Cambrex Charles City, Inc. | Methods and compositions for preparation of amphetamine conjugates and salts thereof |
| WO2017003721A1 | Jun 17, 2016 | Jan 5, 2017 | Noramco, Inc. | Process for the preparation of lisdexamfetamine and related derivatives |
References
- Jump up^ "Public Assessment Report Decentralised Procedure" (PDF). Shire Pharmaceuticals Contracts Limited. p. 14. Retrieved 23 August 2014.
- ^ Jump up to:a b Millichap JG (2010). "Chapter 9: Medications for ADHD". In Millichap JG. Attention Deficit Hyperactivity Disorder Handbook: A Physician's Guide to ADHD (2nd ed.). New York, USA: Springer. p. 112. ISBN 9781441913968.
Table 9.2 Dextroamphetamine formulations of stimulant medication
Dexedrine [Peak:2–3 h] [Duration:5–6 h] ...
Adderall [Peak:2–3 h] [Duration:5–7 h]
Dexedrine spansules [Peak:7–8 h] [Duration:12 h] ...
Adderall XR [Peak:7–8 h] [Duration:12 h]
Vyvanse [Peak:3–4 h] [Duration:12 h] - ^ Jump up to:a b Brams M, Mao AR, Doyle RL (September 2008). "Onset of efficacy of long-acting psychostimulants in pediatric attention-deficit/hyperactivity disorder". Postgrad. Med. 120 (3): 69–88. PMID 18824827. doi:10.3810/pgm.2008.09.1909.
Onset of efficacy was earliest for d-MPH-ER at 0.5 hours, followed by d, l-MPH-LA at 1 to 2 hours, MCD at 1.5 hours, d, l-MPH-OR at 1 to 2 hours, MAS-XR at 1.5 to 2 hours, MTS at 2 hours, and LDX at approximately 2 hours. ... MAS-XR, and LDX have a long duration of action at 12 hours postdose
- ^ Jump up to:a b c d e f g h i j k l m "Vyvanse Prescribing Information" (PDF). United States Food and Drug Administration. Shire US Inc. January 2015. Retrieved 24 February 2015.
- ^ Jump up to:a b Heal DJ, Smith SL, Gosden J, Nutt DJ (June 2013). "Amphetamine, past and present – a pharmacological and clinical perspective". J. Psychopharmacol. 27 (6): 479–496. PMC 3666194
 . PMID 23539642. doi:10.1177/0269881113482532.
. PMID 23539642. doi:10.1177/0269881113482532. - Jump up^ "Lisdexamfetamine dimesylate (generic)." Brown University Psychopharmacology Update 19.7 (2008): 1–2. Academic Search Premier. EBSCO. Web. 12 September 2010.
- Jump up^ "DEA – Department of Justice" (PDF). DEA – Department of Justice. Retrieved 1 July 2014.
- Jump up^ "DEA Office of Diversion Control" (PDF). DEA. Retrieved 1 July 2014.
- Jump up^ "Phase-III clinical trials in Attention-deficit hyperactivity disorder (In children, In adolescents) in Japan (PO)". Retrieved 20 March 2016.
- ^ Jump up to:a b Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remião F, Carvalho F, Bastos Mde L (August 2012). "Toxicity of amphetamines: an update". Arch. Toxicol. 86 (8): 1167–1231. PMID 22392347. doi:10.1007/s00204-012-0815-5.
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Tyvense, Elvanse, Venvanse, Vyvanse |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607047 |
| License data |
|
| Pregnancy category | |
| Dependence liability | Physical: none Psychological: moderate |
| Addiction liability | Moderate |
| Routes of administration | Oral (capsules) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 96.4%[1] |
| Metabolism | Hydrolysis by enzymes in red blood cells initially. Subsequent metabolism follows Amphetamine#Pharmacokinetics. |
| Onset of action | 2 hours[2][3] |
| Biological half-life | ≤1 hour (prodrug molecule) 9–11 hours (dextroamphetamine) |
| Duration of action | 12 hours[2][3] |
| Excretion | Renal: ~2% |
| Identifiers | |
| Synonyms | Vyvanse |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H25N3O |
| Molar mass | 263.378 g/mol |
| 3D model (Jmol) | |
////////
CC(CC1=CC=CC=C1)NC(=O)C(CCCCN)N