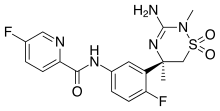
Verubecestat
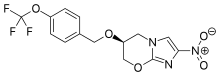
Verubecestat Impurity 10
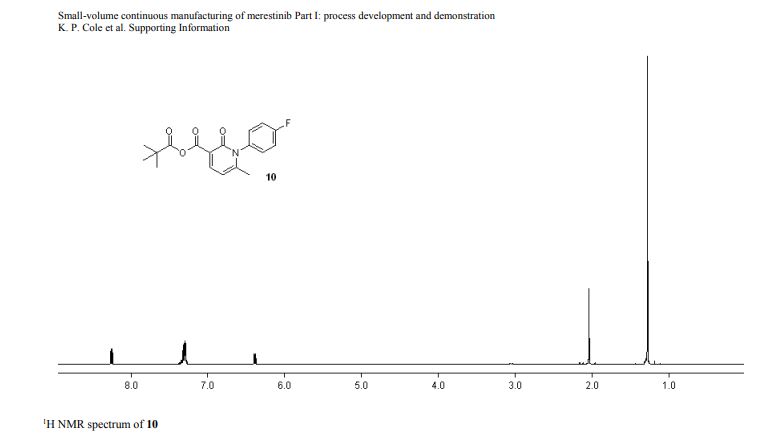
1H NMR (600 MHz, Acetonitrile-d3) δ 9.86 (s, 1H), 8.21 (d, J = 3.0 Hz, 1H), 8.02 (dd, J = 7.5, 2.8 Hz, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.81 (ddd, J = 8.7, 4.2, 2.7 Hz, 1H), 7.24 (dd, J = 8.9, 3.0 Hz, 1H), 7.19 (d, J = 8.7 Hz, 2H), 7.07 (dd, J = 12.0, 8.7 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 4.06 (d, J = 14.6 Hz, 1H), 3.92 (d, J = 14.6 Hz, 1H), 3.81 (d, J = 14.2 Hz, 1H), 3.75 (s, 3H), 3.65 (ddd, J = 11.7, 9.8, 3.9 Hz, 1H), 3.39 (d, J = 14.3 Hz, 1H), 2.86 (s, 3H), 2.59 (td, J = 10.3, 4.0 Hz, 1H), 2.55 (s, 3H), 2.28 (s, 3H), 2.22 – 2.09 (m, 1H), 1.78 – 1.71 (m, 2H), 1.69 – 1.64 (m, 1H), 1.61 – 1.54 (m, 4H), 1.41 (qt, J = 13.7, 3.9 Hz, 1H), 1.28 (qt, J = 13.3, 3.8 Hz, 1H), 1.17 – 1.07 (m, 1H).
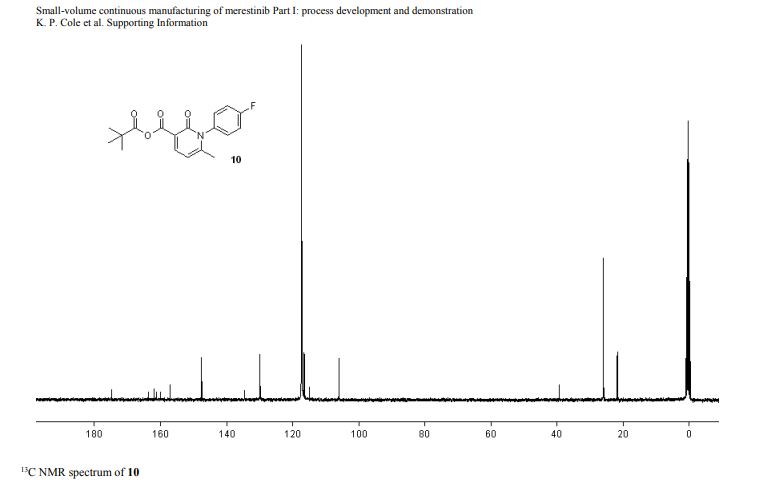
13C NMR (151 MHz, Acetonitrile-d3) δ 163.79, 160.26, 156.68, 149.74, 137.95, 136.00 (d, J = 2.3Hz), 134.69 (d, J = 13.7 Hz), 134.15, 130.58, 129.35, 123.67, 120.82 (d, J = 8.7 Hz), 120.51 (d, J = 4.3 Hz), 119.60, 116.85, 114.82, 63.09, 60.77, 60.31 (d, J = 5.5 Hz), 55.84, 54.27 (d, J = 3.4 Hz), 53.30, 34.11, 33.75, 31.85, 30.96, 30.23 (d, J = 3.3 Hz), 28.91, 26.13, 25.25.
19F NMR (564 MHz, Acetonitrile-d3) δ -119.08. HRMS [M+H] (C32H43FN6O4S) calc. 627.3123, obs. 627.3151.
Improved Process for a Copper-Catalyzed C–N Coupling in the Synthesis of Verubecestat
Eric M. Phillips*
Cite This:Org. Process Res. Dev.2019XXXXXXXXXX-XXX
Publication Date:July 23, 2019
Verubecestat is a β-site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitor which was previously evaluated for the treatment of Alzheimer’s disease. The synthesis of verubecestat relies on a Cu-catalyzed carbon–nitrogen coupling. During process development, observations of impurity formation led to a more robust understanding of the catalyst. The transformation was discovered to be highly dependent on the ratio of ligand to substrate concentration during the course of the reaction. In-depth studies aimed at attaining mechanistic understanding provided an explanation of experimental findings and ultimately led to the identification of conditions that resulted in a more robust process.
///////////Verubecestat, Impurity 10

 *a
*a 
























 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..