
Migalastat hydrochloride
CAS Number: 75172-81-5 hydrochloride
CAS Number: 75172-81-5 hydrochloride
CAS BASE....108147-54-2
ABS ROT = (+)
| Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride (1:1), (2R,3S,4R,5S)-
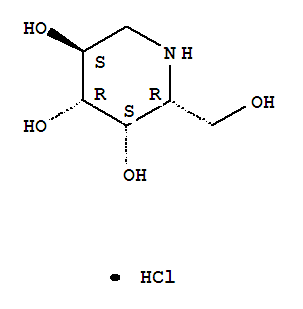
Molecular Structure:

Formula: C6H14ClNO4
Molecular Weight:199.63
Synonyms: 3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);
Formula: C6H14ClNO4
Molecular Weight:199.63
Synonyms: 3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, (2R,3S,4R,5S)- (9CI);
3,4,5-Piperidinetriol,2-(hydroxymethyl)-, hydrochloride, [2R-(2a,3a,4a,5b)]-;
Migalastat hydrochloride;Galactostatin hydrochloride;
(2S,3R,4S,5S)-2-(hydroxymethyl)piperidine-3,4,5-triol hydrochloride;
- 1-Deoxygalactonojirimycin
- 1-Deoxygalactostatin
- Amigal
- DDIG
- Migalastat
- UNII-C4XNY919FW
Melting Point:160-2 °C.........http://www.google.com/patents/DE3906463A1?cl=de
Boiling Point:382.7 °C at 760 mmHg
Flash Point:185.2 °C
Boiling Point:382.7 °C at 760 mmHg
Flash Point:185.2 °C
Amicus Therapeutics, Inc. innovator

Aug 2014
Amicus Therapeutics was on the ropes in late 2012 when its pill for a rare condition called Fabry Disease108147-54-2 failed a late-stage trial. It had already put seven years of work into the drug, and the setback added even more development time and uncertainty to the mix. But the Cranbury, NJ-based company kept plugging away, and now it looks like all the effort could lead to its first approved drug.
Amicus (NASDAQ: FOLD) is reporting today that the Fabry drug, migalastat, succeeded in the second of two late-stage trials. It hit two main goals that essentially measured its ability to slow the decline of Fabry patients’ kidney function comparably to enzyme-replacement therapy (ERT)—the standard of care for the often-fatal disorder.
Amicus believes the results, along with those from an earlier Phase 3 trial comparing migalastat to a placebo, are good enough to ask regulators in the U.S. and Europe for market approval.
“These are the good days to be a CEO,” says Amicus CEO John Crowley (pictured above). “It’s great when a plan comes together and data cooperates.”
Crowley says Amicus will seek approval of migalastat first in Europe and is already in talks with regulators there. In the next few months, Amicus will begin talking with the FDA about a path for approval in the U.S. as well.
End feb 2013
About Amicus Therapeutics
Amicus Therapeutics is a biopharmaceutical company at the forefront of therapies for rare and orphan diseases. The Company is developing orally-administered, small molecule drugs called pharmacological chaperones, a novel, first-in-class approach to treating a broad range of human genetic diseases. Amicus’ late-stage programs for lysosomal storage disorders include migalastat HCl monotherapy in Phase 3 for Fabry disease; migalastat HCl co-administered with enzyme replacement therapy (ERT) in Phase 2 for Fabry disease; and AT2220 co-administered with ERT in Phase 2 for Pompe disease.
About Migalastat HCl
Amicus in collaboration with GlaxoSmithKline (GSK) is developing the investigational pharmacological chaperone migalastat HCl for the treatment of Fabry disease. Amicus has commercial rights to all Fabry products in the United States and GSK has commercial rights to all of these products in the rest of world.
As a monotherapy, migalastat HCl is designed to bind to and stabilize, or “chaperone” a patient’s own alpha-galactosidase A (alpha-Gal A) enzyme in patients with genetic mutations that are amenable to this chaperone in a cell-based assay. Migalastat HCl monotherapy is in Phase 3 development (Study 011 and Study 012) for Fabry patients with genetic mutations that are amenable to this chaperone monotherapy in a cell-based assay. Study 011 is a placebo-controlled study intended primarily to support U.S. registration, and Study 012 compares migalastat HCl to ERT to primarily support global registration.
For patients currently receiving ERT for Fabry disease, migalastat HCl in combination with ERT may improve ERT outcomes by keeping the infused alpha-Gal A enzyme in its properly folded and active form thereby allowing more active enzyme to reach tissues.2Migalastat HCl co-administered with ERT is in Phase 2 (Study 013) and migalastat HCl co-formulated with JCR Pharmaceutical Co. Ltd’s proprietary investigational ERT (JR-051, recombinant human alpha-Gal A enzyme) is in preclinical development.
About Fabry Disease
Fabry disease is an inherited lysosomal storage disorder caused by deficiency of an enzyme called alpha-galactosidase A (alpha-Gal A). The role of alpha-Gal A within the body is to break down specific lipids in lysosomes, including globotriaosylceramide (GL-3, also known as Gb3). Lipids that can be degraded by the action of α-Gal are called “substrates” of the enzyme. Reduced or absent levels of alpha-Gal A activity leads to the accumulation of GL-3 in the affected tissues, including the kidneys, heart, central nervous system, and skin. This accumulation of GL-3 is believed to cause the various symptoms of Fabry disease, including pain, kidney failure, and increased risk of heart attack and stroke.
It is currently estimated that Fabry disease affects approximately 5,000 to 10,000 people worldwide. However, several literature reports suggest that Fabry disease may be significantly under diagnosed, and the prevalence of the disease may be much higher.
2. Benjamin, et al., Molecular Therapy: April 2012, Vol. 20, No. 4, pp. 717–726.
Migalastat hydrochloride is a pharmacological chaperone in phase III development at Amicus Pharmaceuticals for the oral treatment of Fabry's disease. Fabry's disease occurs as the result of an inherited genetic mutation that results in the production of a misfolded alpha galactosidase A (alpha-GAL) enzyme, which is responsible for breaking down globotriaosylceramide (GL-3) in the lysosome. Migalastat acts by selectively binding to the misfolded alpha-GAL, increasing its stability and promoting proper folding, processing and trafficking of the enzyme from the endoplasmic reticulum to the lysosome.
In February 2004, migalastat hydrochloride was granted orphan drug designation by the FDA for the treatment of Fabry's disease.
The EMEA assigned orphan drug designation for the compound in 2006 for the treatment of the same indication. In 2007, the compound was licensed to Shire Pharmaceuticals by Amicus Therapeutics worldwide, with the exception of the U.S., for the treatment of Fabry's disease.
In 2009, this license agreement was terminated. In 2010, the compound was licensed by Amicus Therapeutics to GlaxoSmithKline on a worldwide basis to develop, manufacture and commercialize migalastat hydrochloride as a treatment for Fabry's disease, but the license agreement terminated in 2013.
| Chemical Name: | DEOXYGALACTONOJIRIMYCIN, HYDROCHLORIDE |
| Synonyms: | DGJ;Amigal;Unii-cly7m0xd20;GALACTOSTATIN HCL;DGJ, HYDROCHLORIDE;Migalastat hydrochloride;Galactostatin hydrochloride;DEOXYGALACTONOJIRIMYCIN HCL;1-DEOXYGALACTONOJIRIMYCIN HCL;1,5-dideoxy-1,5-imino-d-galactitol |

.............................

Example 1
A series of plant alkaloids (Scheme 1, ref. 9) were used for both in vitro inhibition and intracellular enhancement studies of α-Gal A activity. The results of inhibition experiments are shown in Fig. 1 A.

f^

Among the tested compounds, 1-deoxy-galactonojirimycin (DGJ, 5) known as a powerful competitive inhibitor for α-Gal A, showed the highest inhibitory activity with IC50 at 4.7 nM. α-3,4-Di-epi-homonojirimycin (3) was an effective inhibitor with IC50 at 2.9 μM. Other compounds showed moderate inhibitory activity with IC50 ranging from 0.25 mM (6) to 2.6 mM (2). Surprisingly, these compounds also effectively enhanced α-Gal A activity in COS-1 cells transfected with a mutant α-Gal A gene (R301Q), identified from an atypical variant form of Fabry disease with a residual α- Gal A activity at 4% of normal. By culturing the transfected COS-1 cells with these compounds at concentrations cat 3 - 10-fold of IC50 of the inhibitors, α-Gal A activity was enhanced 1.5 - 4-fold (Fig. 1C). The effectiveness of intracellular enhancement paralleled with in vitro inhibitory activity while the compounds were added to the culture medium at lOμM
concentration (Fig. IB).
...........................
WO 2008045015
This invention relates to a process for purification of imino or amino sugars, such as D-1-deoxygalactonojirimycin hydrochloride (DGJ'HCl). This process can be used to produce multi-kilogram amounts of these nitrogen-containing sugars.
Sugars are useful in pharmacology since, in multiple biological processes, they have been found to play a major role in the selective inhibition of various enzymatic functions. One important type of sugars is the glycosidase inhibitors, which are useful in treatment of metabolic disorders. Galactosidases catalyze the hydrolysis of glycosidic linkages and are important in the metabolism of complex carbohydrates. Galactosidase inhibitors, such as D-I- deoxygalactonojirimycin (DGJ), can be used in the treatment of many diseases and conditions, including diabetes (e.g., U.S. Pat. 4,634,765), cancer (e.g., U.S. Pat. 5,250,545), herpes (e.g. , U.S. Pat. 4,957,926), HIV and Fabry Disease (Fan et al, Nat. Med. 1999 5:1, 112-5).
Commonly, sugars are purified through chromatographic separation. This can be done quickly and efficiently for laboratory scale synthesis, however, column chromatography and similar separation techniques become less useful as larger amounts of sugar are purified. The size of the column, amount of solvents and stationary phase (e.g. silica gel) required and time needed for separation each increase with the amount of product purified, making purification from multi-kilogram scale synthesis unrealistic using column chromatography.
Another common purification technique for sugars uses an ion- exchange resin. This technique can be tedious, requiring a tedious pre-treatment of the ion exchange resin. The available ion exchange resins are also not necessarily able to separate the sugars from salts (e.g., NaCl). Acidic resins tend to remove both metal ions found in the crude product and amino- or imino-sugars from the solution and are therefore not useful. Finding a resin that can selectively remove the metal cations and leave amino- or imino-sugars in solution is not trivial. In addition, after purification of a sugar using an ion exchange resin, an additional step of concentrating the diluted aqueous solution is required. This step can cause decomposition of the sugar, which produces contaminants, and reduces the yield.
U.S. Pats. 6,740,780, 6,683,185, 6,653,482, 6,653,480, 6,649,766, 6,605,724, 6,590,121, and 6,462,197 describe a process for the preparation of imino- sugars. These compounds are generally prepared from hydroxyl-protected oxime intermediates by formation of a lactam that is reduced to the hexitol. However, this process has disadvantages for the production on a multi-kg scale with regard to safety, upscaling, handling, and synthesis complexity. For example, several of the disclosed syntheses use flash chromatography for purification or ion-exchange resin treatment, a procedure that is not practicable on larger scale. One particularly useful imino sugar is DGJ. There are several DGJ preparations disclosed in publications, most of which are not suitable for an industrial laboratory on a preparative scale (e.g., >100 g). One such synthesis include a synthesis from D-galactose (Santoyo-Gonzalez, et al, Synlett 1999 593-595; Synthesis 1998 1787-1792), in which the use of chromatography is taught for the purification of the DGJ as well as for the purification of DGJ intermediates. The use of ion exchange resins for the purification of DGJ is also disclosed, but there is no indication of which, if any, resin would be a viable for the purification of DGJ on a preparative scale. The largest scale of DGJ prepared published is 13 g (see Fred-Robert Heiker, Alfred Matthias Schueller, Carbohydrate Research, 1986, 119-129). In this publication, DGJ was isolated by stirring with ion-exchange resin Lewatit MP 400 (OH") and crystallized with ethanol. However, this process cannot be readily scaled to multi- kilogram quantities.
Similarly, other industrial and pharmaceutically useful sugars are commonly purified using chromatography and ion exchange resins that cannot easily be scaled up to the purification of multi-kilogram quantities.
Therefore, there is a need for a process for purifying nitrogen- containing sugars, preferably hexose amino- or imino-sugars that is simple and cost effective for large-scale synthesis
FIG. 1. HPLC of purified DGJ after crystallization. The DGJ is over 99.5% pure.

FIG. 2A. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 - 15 ppm in DMSO.

FIG. 2B. 1H NMR of DGJ (post HCl extraction and crystallization), from 0 - 5 ppm, in DMSO.

FIG. 3 A. 1H NMR of purified DGJ (after recrystallization), from 0 - 15 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 3B. 1H NMR of purified DGJ (after recrystallization), from 0 -
4 ppm, in D2O. Note OH moiety has exchanged with OD.

FIG. 4. 13C NMR of purified DGJ, (after recrystallization), 45 - 76 ppm.

One amino-sugar of particular interest for purification by the method of the current invention is DGJ. DGJ, or D-l-deoxygalactonojirimycin, also described as (2R,3S,4R,5S)-2-hydroxymethyl-3,4,5-trihydroxypiperidine and 1- deoxy-galactostatin, is a noj irimycin (5-amino-5-deoxy-D-galactopyranose) derivative of the form:

Example 1: Preparation and Purification of DGJ
A protected crystalline galactofuranoside obtained from the technique described by Santoyo-Gonzalez. 5-azido-5-deoxy-l,2,3,6-tetrapivaloyl-α-D- galactofuranoside (1250 g), was hydrogenated for 1-2 days using methanol (10 L) with palladium on carbon (10%, wet, 44 g) at 50 psi of H2. Sodium methoxide (25% in methanol, 1.25 L) was added and hydrogenation was continued for 1-2 days at 100 psi ofH2. Catalyst was removed by filtration and the reaction was acidified with methanolic hydrogen chloride solution (20%, 1.9 L) and concentrated to give crude mixture of DGJ • HCl and sodium chloride as a solid. The purity of the DGJ was about 70% (w/w assay), with the remaining 30% being mostly sodium chloride.
The solid was washed with tetrahydrofuran (2 x 0.5 L) and ether (I x 0.5 L), and then combined with concentrated hydrochloric acid (3 L). DGJ went into solution, leaving NaCl undissolved. The obtained suspension was filtered to remove sodium chloride; the solid sodium chloride was washed with additional portion of hydrochloric acid (2 x 0.3 L). All hydrochloric acid solution were combined and slowly poured into stirred solution of tetrahydrofuran (60 L) and ether (11.3 L). The precipitate formed while the stirring was continued for 2 hours. The solid crude DGJ* HCl, was filtered and washed with tetrahydrofuran (0.5 L) and ether (2 x 0.5 L). An NMR spectrum is shown in FIGS. 2A-2B.
The solid was dried and recrystallized from water (1.2 mL /g) and ethanol (10 ml/1 ml of water). This recrystallization step may be repeated. This procedure gave white crystalline DGJ* HCl, and was usually obtained in about 70- 75% yield (320 - 345 g). The product of the purification, DGJ-HCl is a white crystalline solid, HPLC >98% (w/w assay) as shown in FIG. 1. FIGS. 3A-3D and FIG. 4 show the NMR spectra of purified DGJ, showing the six sugar carbons.
Example 2: Purification of 1-deoxymannojirimycin 1 -deoxymannojirimycin is made by the method described by Mariano
(J. Org. Chem., 1998, 841-859, see pg. 859, herein incorporated by reference). However, instead of purification by ion-exchange resin as described by Mariano, the 1-deoxymannojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the 1-deoxymannojirimycin hydrochloride is precipitated crystallized using solvents known for recrystallization of 1- deoxymannojirimycin (THF for crystallization and then ethanol/water.
Example 3: Purification of (+)-l-deoxynojirimycin
(+)-l-deoxynojirimycin is made by the method Kibayashi et al. (J. Org. Chem., 1987, 3337-3342, see pg. 334I5 herein incorporated by reference). It is synthesized from a piperidine compound (#14) in HCl/MeOH. The reported yield of 90% indicates that the reaction is essentially clean and does not contain other sugar side products. Therefore, the column chromatography used by Kibayashi is for the isolation of the product from non-sugar related impurities. Therefore, instead of purification by silica gel chromatography, the (+)-l-deoxynojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the salt and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
Example 4: Purification of Nojirimycin
Nojirimycin is made by the method described by Kibayashi et al. (J.
Org. Chem., 1987, 3337-3342, see pg. 3342). However, after evaporating of the mixture at reduced pressure, instead of purification by silica gel chromatography with ammonia-methanol-chloroform as described by Kibayashi, the nojirimycin is mixed with concentrated HCl. The suspension is then filtered to remove the impurities not dissolved in HCl and the nojirimycin is crystallized using solvents known for recrystallization of nojirimycin.
............................
Synthesis of (+)-1-deoxygalactonojirimycin and a related indolizidine
Tetrahedron Lett 1995, 36(5): 653
Tetrahedron Lett 1995, 36(5): 653
Synthesis of (+)-1-deoxygalactonojirimycin and a related indolizidine
Original Research Article- Pages 653-654
- Carl R. Johnson, Adam Golebiowski, Hari Sundram, Michael W. Miller, Renee L. Dwaihy
- Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and indolizidine 4.

Amido-alcohol 1 is transformed via aminal 2 into 1-deoxygalactonojirimycin (3) and the structurally related indolizidine 4.
...........................
Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1,5-imino-D-galactitol) starting from 1-deoxynojirimycin
Carbohydr Res 1990, 203(2): 314
Carbohydr Res 1990, 203(2): 314
Synthesis of d-galacto-1-deoxynojirimycin (1,5-dideoxy-1,5-imino-d-galactitol) starting from 1-deoxynojirimycin
- Pages 314-318
- Fred-Robert Heiker, Alfred Matthias Schueller
......................................
Synthesis of (+)-1,5-dideoxy-1,5-imino-D-galactitol, a potent alpha-D-galactosidase inhibitor
Carbohydr Res 1987, 167: 305
Carbohydr Res 1987, 167: 305
Synthesis of (+)-1,5-dideoxy-1,5-imino-d-galactitol, a potent α-d-galactosidase inhibitor
- Pages 305-311
- Ronald C. Bernotas, Michael A. Pezzone, Bruce Ganem
...................................
SEE
Monosaccharides containing nitrogen in the ring, XXXVII. Synthesis of 1,5-didexy-1,5-imino-D-galactitol
Chem Ber 1980, 113(8): 2601
Chem Ber 1980, 113(8): 2601
..............................
Org. Lett., 2010, 12 (17), pp 3957–3959
DOI: 10.1021/ol101556k
| Conc: 1 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C |
IN

The chemoenzymatic synthesis of three 1-deoxynojirimycin-type iminosugars is reported. Key steps in the synthetic scheme include a Dibal reduction−transimination−sodium borohydride reduction cascade of reactions on an enantiomerically pure cyanohydrin, itself prepared employing almond hydroxynitrile lyase (paHNL) as the common precursor. Ensuing ring-closing metathesis and Upjohn dihydroxylation afford the target compounds.
COMPD 18
D-galacto-1-deoxynojirimicin.HCl (18).
D-N-Boc-6-OBn-galacto-1-deoxynojirimicin (159 mg, 0.450 mmol) was dissolved in a mixture of MeOH
(10 mL) and 6 M HCl (2 mL). The flask was purged with argon, Pd/C-10% (20 mg) was added and a balloon
with hydrogen gas was placed on top of the reaction. The mixture was stirred overnight at room temperature.
Pd/C was removed by filtration and the filtrate evaporated to yield the crude product (90 mg, 100%) as a
white foam that needed no further purification.
[α]24D = + 53.0 (c = 1, H2O);
(10 mL) and 6 M HCl (2 mL). The flask was purged with argon, Pd/C-10% (20 mg) was added and a balloon
with hydrogen gas was placed on top of the reaction. The mixture was stirred overnight at room temperature.
Pd/C was removed by filtration and the filtrate evaporated to yield the crude product (90 mg, 100%) as a
white foam that needed no further purification.
[α]24D = + 53.0 (c = 1, H2O);
[lit4a [α]24D = +44.6 (c = 0.9, H2O); lit4b [α]20D = +46.1 (c = 0.9, H2O)].
HRMS calculated for [C6H13NO4 + H]+164.09173; Found 164.09160.
1H NMR (400 MHz, D2O) δ 4.20 (dd, J = 2.7, 1.1 Hz, 1H), 4.11 (ddd, J = 11.4, 9.7, 5.4 Hz, 1H), 3.88 (ddd,
J = 20.9, 12.2, 6.8 Hz, 2H), 3.68 (dd, J = 9.7, 3.0 Hz, 1H), 3.55 (dd, J = 12.5, 5.4 Hz, 1H), 3.46 (ddd, J = 8.6,
4.8, 1.0 Hz, 1H), 2.97 – 2.86 (t, J = 12.0 Hz, 1H). [lit4c supporting information contains 1
H NMR-spectrumof an authentic sample].
13C NMR (101 MHz, D2O) δ 73.01, 66.97, 64.69, 60.16, 59.15, 46.15
HRMS calculated for [C6H13NO4 + H]+164.09173; Found 164.09160.
1H NMR (400 MHz, D2O) δ 4.20 (dd, J = 2.7, 1.1 Hz, 1H), 4.11 (ddd, J = 11.4, 9.7, 5.4 Hz, 1H), 3.88 (ddd,
J = 20.9, 12.2, 6.8 Hz, 2H), 3.68 (dd, J = 9.7, 3.0 Hz, 1H), 3.55 (dd, J = 12.5, 5.4 Hz, 1H), 3.46 (ddd, J = 8.6,
4.8, 1.0 Hz, 1H), 2.97 – 2.86 (t, J = 12.0 Hz, 1H). [lit4c supporting information contains 1
H NMR-spectrumof an authentic sample].
13C NMR (101 MHz, D2O) δ 73.01, 66.97, 64.69, 60.16, 59.15, 46.15
4a) Ruiz, M.; Ruanova, T. M.; Blanco, O.; Núñez, F.; Pato, C.; Ojea, V. J. Org. Chem. 2008, 73, 2240
– 2255.
– 2255.
4b) Paulsen, H.; Hayauchi, Y.; Sinnwell, V. Chem. Ber. 1980, 113, 2601 – 2608. c)
McDonnell, C.; Cronin, L.; O’Brien, J. L.; Murphy, P. V. J. Org. Chem. 2004, 69, 3565 – 3568.
McDonnell, C.; Cronin, L.; O’Brien, J. L.; Murphy, P. V. J. Org. Chem. 2004, 69, 3565 – 3568.
..............................................
(- ) FORM............ BE CAREFUL
Short and straightforward synthesis of (-)-1-deoxygalactonojirimycin
Org Lett 2010, 12(6): 1145
Org Lett 2010, 12(6): 1145

The mildness and low basicity of vinylzinc species functioning as a nucleophile in addition to α-chiral aldehydes is characterized by lack of epimerization of the vulnerable stereogenic center. This is demonstrated by a highly diastereoselective synthesis of 1-deoxygalactonojirimycin in eight steps from commercial starting materials with overall yield of 35%.

Figure 1. Structures of nojirimycin (1) and DGJ (2).
SEE SUPP INFO
(-)-1-deoxygalactojirimycin hydrochloride as transparent colorless needles.
[α]D -51.4 (D2O, c 1.0)
[α]D -51.4 (D2O, c 1.0)
1H-NMR (D2O) δ ppm 4.09 (dd, 1H, J 2.9 Hz, 1.3 Hz), 4.00 (ddd, 1H, J = 11.3 Hz, 9.7 Hz, 5.3 Hz),
3.80 (dd, 1H, J = 12,1 Hz, 8.8 Hz), 3.73 (dd, 1H, J = 12.1 Hz, 8.8 Hz), 3.56 (dd, 1H, J = 9.7 Hz, 2.9
Hz), 3.44 (dd, 1H, J = 12.4 Hz, 5.3 Hz), 3.34 (ddd, 1H, J = 8.7 Hz, 4.8 Hz, 1.0 Hz), 2.8 (app. t, 1H,
J = 12.0 Hz)
13C-NMR (D2O, MeOH iSTD) δ 73.6, 67.5, 65.3, 60.7, 59.7, 46.7
HRMS Measured 164.0923 (M + H - Cl) Calculated 164.0923 (C6H13NO4 + H - Cl)
3.80 (dd, 1H, J = 12,1 Hz, 8.8 Hz), 3.73 (dd, 1H, J = 12.1 Hz, 8.8 Hz), 3.56 (dd, 1H, J = 9.7 Hz, 2.9
Hz), 3.44 (dd, 1H, J = 12.4 Hz, 5.3 Hz), 3.34 (ddd, 1H, J = 8.7 Hz, 4.8 Hz, 1.0 Hz), 2.8 (app. t, 1H,
J = 12.0 Hz)
13C-NMR (D2O, MeOH iSTD) δ 73.6, 67.5, 65.3, 60.7, 59.7, 46.7
HRMS Measured 164.0923 (M + H - Cl) Calculated 164.0923 (C6H13NO4 + H - Cl)
...........................................................
Concise and highly stereocontrolled synthesis of 1-deoxygalactonojirimycin and its congeners using dioxanylpiperidene, a promising chiral building block
Org Lett 2003, 5(14): 2527
Org Lett 2003, 5(14): 2527

A concise and stereoselective synthesis of the chiral building block, dioxanylpiperidene 4 as a precursor for deoxyazasugars, starting from the Garner aldehyde 5 using catalytic ring-closing metathesis (RCM) for the construction of the piperidine ring is described. The asymmetric synthesis of 1-deoxygalactonojirimycin and its congeners 1−3 was carried out via the use of 4in a highly stereocontrolled mode.
mp 135-135.5 °C [lit.3mp 137-139 °C];
[α]D25 +27.8° (c 0.67, H2O)
[lit.3[α]D23 +28° (c 0.5, H2O)];
[lit.3[α]D23 +28° (c 0.5, H2O)];
1H NMR (300 MHz, D2O) δ 2.59–2.65 (m, 1H), 2.81–2.87 (m, 1H),
3.02–3.08 (m, 1H), 3.46–3.48 (m, 2H), 3.59–3.66 (m, 3H); 13C NMR (75 MHz, D2O) δ 44.7, 57.1,
3.02–3.08 (m, 1H), 3.46–3.48 (m, 2H), 3.59–3.66 (m, 3H); 13C NMR (75 MHz, D2O) δ 44.7, 57.1,
58.4, 70.9, 71.4, 73.3 [lit4 13C NMR (125 MHz, D2O) δ 44.5, 56.8, 58.3, 70.1, 70.7, 72.3];
HRMScalcd for C6H13NO4 (M+) 163.0855, Found 163.0843. Anal. calcd for C6H13NO4: C, 44.16; N,
8.58; H, 8.03. Found: C, 44.31; N, 8.55; H, 7.71.
8.58; H, 8.03. Found: C, 44.31; N, 8.55; H, 7.71.
3. Schaller, C.; Vogel, P.; Jager, V. Carbohydrate Res. 1998, 314, 25-35.
4. Lee, B. W.; Jeong, Ill-Y.; Yang, M. S.; Choi, S. U.; Park, K. H. Synthesis 2000, 1305-1309.
4. Lee, B. W.; Jeong, Ill-Y.; Yang, M. S.; Choi, S. U.; Park, K. H. Synthesis 2000, 1305-1309.
..................................................
Applications and limitations of the I2-mediated carbamate annulation for the synthesis of piperidines: Five- versus six-membered ring formation
J Org Chem 2013, 78(19): 9791
J Org Chem 2013, 78(19): 9791

A protecting-group-free synthetic strategy for the synthesis of piperidines has been explored. Key in the synthesis is an I2-mediated carbamate annulation, which allows for the cyclization of hydroxy-substituted alkenylamines into piperidines, pyrrolidines, and furans. In this work, four chiral scaffolds were compared and contrasted, and it was observed that with both d-galactose and 2-deoxy-d-galactose as starting materials, the transformations into the piperidines 1-deoxygalactonorjirimycin (DGJ) and 4-epi-fagomine, respectively, could be achieved in few steps and good overall yields. When d-glucose was used as a starting material, only the furan product was formed, whereas the use of 2-deoxy-d-glucose resulted in reduced chemo- and stereoselectivity and the formation of four products. A mechanistic explanation for the formation of each annulation product could be provided, which has improved our understanding of the scope and limitations of the carbamate annulation for piperidine synthesis.
...................................................
ROT +44.6 ° Conc: 0.9 g/100mL; Solv: water ; 589.3 nm; Temp: 24 °C

A general strategy for the synthesis of 1-deoxy-azasugars from a chiral glycine equivalent and 4-carbon building blocks is described. Diastereoselective aldol additions of metalated bislactim ethers to matched and mismatched erythrose or threose acetonides and intramolecular N-alkylation (by reductive amination or nucleophilic substitution) were used as key steps. The dependence of the yield and the asymmetric induction of the aldol addition with the nature of the metallic counterion of the azaenolate and the γ-alkoxy protecting group for the erythrose or threose acetonides has been studied. The stereochemical outcome of the aldol additions with tin(II) azaenolates has been rationalized with the aid of density functional theory (DFT) calculations. In accordance with DFT calculations with model glyceraldehyde acetonides, hightrans,syn,anti-selectivitity for the matched pairs and moderate to low trans,anti,anti-selectivity for the mismatched ones may originate from (1) the intervention of solvated aggregates of tin(II) azaenolate and lithium chloride as the reactive species and (2) favored chair-like transition structures with a Cornforth-like conformation for the aldehyde moiety. DFT calculations indicate that aldol additions to erythrose acetonides proceed by an initial deprotonation, followed by coordination of the alkoxy-derivative to the tin(II) azaenolate and final reorganization of the intermediate complex through pericyclic transition structures in which the erythrose moiety is involved in a seven-membered chelate ring. The preparative utility of the aldol-based approach was demonstrated by application in concise routes for the synthesis of the glycosidase inhibitors 1-deoxy-d-allonojirimycin, 1-deoxy-l-altronojirimycin, 1-deoxy-d-gulonojirimycin, 1-deoxy-d-galactonojirimycin, 1-deoxy-l-idonojirimycin and 1-deoxy-d-talonojirimycin.
.......................
J. Org. Chem., 1991, 56 (2), pp 815–819
DOI: 10.1021/jo00002a057
..................
Example 1 Preparation of 1,5-dideoxy-1,5-imino-D-glucitol hydrobromide
A suspension of 1,5-dideoxy-1,5-imino-D-glucitol (500 g) in isopropanol (2 l) with 48% hydrochloric acid, bromine (620 g). The suspension is stirred for 2 hours at 40 ° C, cooled to 0 ° C and the product isolated by filtration.
Yield: 700 g (93% of theory),
mp: 184 ° C.
mp: 184 ° C.
Example 2 Preparation of 1,5-dideoxy-1,5-imino-D-mannitol hydrobromide
The prepared analogously to Example 1 from 1,5-dideoxy 1,5-imino-D-mannitol and 48% hydrobromic acid.
Yield: 89% of theory;
C₆H₁₄NO₄Br (244.1)
Ber .: C 29.5%; H 5.8%; N 5.7%; Br 32.7%;
vascular .: C 29.8%; H 5.8%; N 5.8%; Br 32.3%.
Ber .: C 29.5%; H 5.8%; N 5.7%; Br 32.7%;
vascular .: C 29.8%; H 5.8%; N 5.8%; Br 32.3%.
Example 3 Preparation of 1,5-dideoxy-1,5-imino-D-Galactitol- hydrochloride
The preparation was carried out analogously to Example 1 from 1,5-dideoxy-1,5-imino-D-galactitol and corresponding mole ratios of 37% hydrochloric acid.
yield: 91% of theory
, mp: 160-162 ° C.
yield: 91% of theory
, mp: 160-162 ° C.
| Amat et al., "Eantioselective Synthesis of 1-deoxy-D-gluonojirimycin From A Phenylglycinol Derived Lactam," Tetrahedron Letters, pp. 5355-5358, 2004. | ||
| 2 | Chernois, "Semimicro Experimental Organic Chemistry," J. de Graff (1958), pp. 31-48. | |
| 3 | Encyclopedia of Chemical Technology, 4th Ed., 1995, John Wiley & Sons, vol. 14: p. 737-741. | |
| 4 | Heiker et al., "Synthesis of D-galacto-1-deoxynojirimycin (1, 5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin." Carbohydrate Research, 203: 314-318, 1990. | |
| 5 | Heiker et al., 1990, "Synthesis of D-galacto-1-deoxynojirimycin (1,5-dideoxy-1, 5-imino-D-galactitol) starting from 1-deoxynojirimycin," Carbohydrate Research, vol. 203: p. 314-318. | |
| 6 | * | Joseph, Carbohydrate Research 337 (2002) 1083-1087. |
| 7 | * | Kinast et al. Angew. Chem. Int. Ed. Engl. 20 (1998), No. 9, pp. 805-806. |
| 8 | * | Lamb, Laboratory Manual of General Chemistry, Harvard University Press, 1916, p. 108. |
| 9 | Linden et al., "1-Deoxynojirimycin Hydrochloride," Acta ChrystallographicaC50, pp. 746-749, 1994. | |
| 10 | Mellor et al., Preparation, biochemical characterization and biological properties of radiolabelled N-alkylated deoxynojirimycins, Biochem. J. Aug. 15, 2002; 366(Pt 1):225-233. | |
| 11 | * | Mills, Encyclopedia of Reagents for Organic Synthesis, Hydrochloric Acid, 2001 John Wily & Sons. |
| 12 | Santoyo-Gonzalez et al., "Use of N-Pivaloyl Imidazole as Protective Reagent for Sugars." Synthesis 1998 1787-1792. | |
| 13 | Schuller et al., "Synthesis of 2-acetamido-1, 2-dideoxy-D-galacto-nojirimycin (2-acetamido-1, 2, 5-trideoxy-1, 5-imino-D-galacitol) from 1-deoxynojirimycin." Carbohydrate Res. 1990; 203: 308-313. | |
| 14 | Supplementary European Search Report dated Mar. 11, 2010 issued in corresponding European Patent Application No. EP 06 77 2888. | |
| 15 | Uriel et al., A Short and Efficient Synthesis of 1,5-dideoxy-1,5-imino-D-galactitol (1-deoxy-D-galactostatin) and 1,5-dideoxy-1,5-dideoxy-1,5-imino-L-altritol (1-deoxy-L-altrostatin) From D-galactose, Synlett (1999), vol. 5, pp. 593-595. |
1-Deoxygalactonojirimycin:
(a) Liguchi, T.; Tajiri, K.; Ninomiya, I.; Naito, T. Tetrahedron2000, 56, 5819−5833.
(b) Mehta, G.; Mohal, N. Tetrahedron Lett. 2000, 41, 5741−5745.
(c) Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. Chem. Commun. 1999, 41−42.
(d) Johnson, C. R.; Golebiowsky, A.; Sundram, H.; Miller, M. W.; Dwaihy, R. L. TetraherdonLett. 1995, 36, 653−654.
(e) Uriel, C.; Santoyo-Gonzalez, F. Synlett 1999, 593−595.
(f) Ruiz, M.; Ruanova, T. M.; Ojea, V.; Quintela, J. M. Tetrahedron Lett. 1999, 40, 2021−2024.
(g) Shilvock, J. P.; Fleet, G. W. J. Synlett 1998, 554−556.
(h) Chida, N.; Tanikawa, T.; Tobe, T.; Ogawa, S. J. Chem. Soc., Chem. Commun. 1994, 1247−1248.
(i) Aoyagi, S.; Fujimaki, S.; Yamazaki, N.; Kibayashi, C. J. Org. Chem. 1991, 56, 815−819.
(j) Kajimoto, T.; Chen, L.; Liu, K. K. C.; Wong, C. H. J. Am. Chem. Soc. 1991, 113, 6678−6680.
(k) Bernotas, R. C.; Pezzone, M. A.; Ganem, B. Carbohydr. Res. 1987, 167, 305−311. 1-Deoxyidonojirimycin:
(l) Singh, O. V.; Han, H. Tetrahedron Lett. 2003, 44, 2387−2391.
(m) Schaller, C.; Vogel, P.; Jager, V. Carbohydr. Res. 1998, 314, 25−35.
(n) Fowler, P. A.; Haines, A. H.; Taylor, R. J. K.; Chrystal, E. J. T.; Gravestock, M. B. Carbohydr. Res. 1993,246 377−381.
(o) Liu, K. K. C.; Kajimoto, T.; Chen, L.; Zhong, Z.; Ichikawa, Y.; Wong, C. H.J. Org. Chem. 1991, 56, 6280−6289. 1-Deoxygulonojirimycin: ref 5l.
(p) Haukaas, M. H.; O'Doherty, G. A. Org. Lett. 2001, 3, 401−404.
(q) Ruiz, M.; Ojea, V.; Ruanova, T. M.; Quintela, J. M. Tetrahedron: Asymmetry 2002, 13, 795−799. (r) Liao, L.-X.; Wang, Z.-M.; Zhang, H.-X.; Zhou, W.-S. Tetrahedron: Asymmetry 1999, 10, 3649−3657.
LATUR, MAHARASHTRA, INDIA
http://en.wikipedia.org/wiki/Latur
| Latur लातूर Lattalur, Ratnapur | |
|---|---|
| City | |

Latur
| |
| Coordinates: 18.40°N 76.56°ECoordinates: 18.40°N 76.56°E | |
| Country | |
| State | Maharashtra |
| Region | Aurangabad Division |
| District | Latur |
| Settled | Possibly 7th century AD |
| Government | |
| • Body | Latur Municipal Corporation |
| • Mayor | Akhtar Shaikh |
| Area[1] | |
| • Total | 117.78 km2(45.48 sq mi) |
| Area rank | 89 |
| Elevation | 515 m (1,690 ft) |
| Population (2011) | |
| • Total | 382,754 |
| • Rank | 89th |
| • Density | 3,200/km2(8,400/sq mi) |
| Demonym | Laturkar |
| Languages | |
| • Official | Marathi |
| Time zone | IST (UTC+5:30) |
| PIN |
|
| Telephone code | 91-2382 |
| Vehicle registration | MH-24 |
| Sex ratio | 923.54 ♀/1000 ♂ |
| Literacy | 89.67 |
| Distance from Mumbai | 497 kilometres (309 mi) E (land) |
| Distance fromHyderabad | 337 kilometres (209 mi) NW (land) |
| Distance fromAurangabad, Maharashtra | 294 kilometres (183 mi) SE (land) |
| Climate | BSh (Köppen) |
| Precipitation | 666 millimetres (26.2 in) |
| Avg. summer temperature | 41 °C (106 °F) |
| Avg. winter temperature | 13 °C (55 °F) |
| http://www.citypopulation.de/world/Agglomerations.html | |




his Is The Famous 'Ganj-Golai' As The Central Place Of The Latur City. There Are 16 Roads Connecting To This Place And Seperate Markets i.e. Jewellers ...

लातूर जिल्हयातील चित्र संग्रह






LATUR AIRPORT
 LATUR AIRPORT
LATUR AIRPORT


2012 Navratri Mahotsav in Latur

SOS Children's Village Latur


Latur, India: Carnival Resort


Ausa Near Latur

Chakur near Latur

Vilasrao Deshmukh's ancestral home at Babhalgaon village in Latur. Machindra Amle



UDGIR: Udgir is one of the most important towns of Latur district. Udgir has a great historical significance. It has witnessed the war between the Marathas ...
 The city of Latur is located in India's welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the
The city of Latur is located in India's welathiest state, Maharashtra. Together with many of the surrounding villages, Latur was all but destroyed in the