ABSRACT
Two new triterpenoid saponins 1 and 2 were isolated from the methanol extract of the roots of Acanthophyllum gypsophiloides Regel. These saponins have quillaic acid or gypsogenin moieties as an aglycon, and both bear similar sets of two oligosaccharide chains, which are 3-O-linked to the triterpenoid part trisaccharide α-L-Arap-(1→3)-[α-D-Galp-(1→2)]-β-D-GlcpA and pentasaccharide β-D-Xylp-(1→3)-β-D-Xylp-(1→3)-α-L-Rhap-(1→2)-[β-D-Quip-(1→4)]-β-D-Fucp connected through an ester linkage to C-28. The structures of the obtained saponins were elucidated by a combination of mass spectrometry and 2D NMR spectroscopy. A study of acute toxicity, hemolytic, anti-inflammatory, immunoadjuvant and antifungal activity was carried out. Both saponins 1 and 2 were shown to exhibit immunoadjuvant properties within the vaccine composition with keyhole limpet hemocyanin-based immunogen. The availability of saponins 1 and 2 as individual pure compounds from the extract of the roots of A. gypsophiloides makes it a prospective source of immunoactive agents.
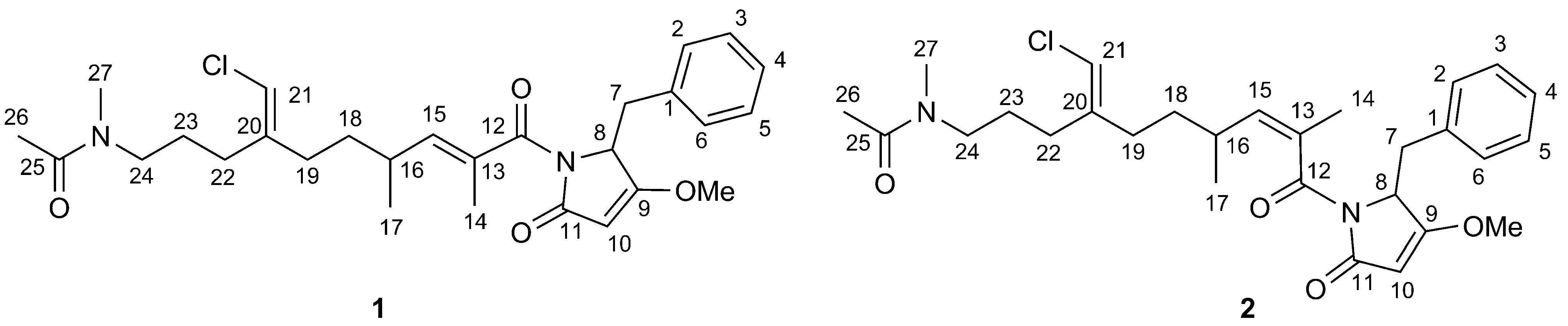
The methanolic extract of the dried powdered roots of A. gypsophiloides was concentrated, and the crude mixture of saponins was precipitated from methanol by the addition of acetone and subjected to reversed-phase С18 HPLC. Compounds 1 and 2 (Figure 1) were isolated as white amorphous powders. Compound 1 exhibited in the HRMS (ESI) the [M + Na]+ peak at m/z 1681.7071, indicating a molecular weight compatible with the molecular formula C75H118O40. Compound 2 exhibited the [M + Na]+ peak at m/z 1665.7181, consistent with the molecular formula С75H118O39. GLC analysis of the acetylated (S)-2-octyl glycosides derived after full acid hydrolysis of compound 1 revealed the presence of D-galactose (D-Gal), L-arabinose (L-Ara), 6-deoxy-D-glucose (D-Qui), D-xylose (D-Xyl), L-rhamnose (L-Rha), D-fucose (D-Fuc), and D-glucuronic acid (D-GlcA). Similar investigation of compound 2 revealed the same sugar composition as for compound 1.
![[1860-5397-8-87-1]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-8-87-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1:
Saponins from A. gypsophiloides 1, R = OH and 2, R = H.
The structures of both compounds 1 and 2 were confirmed on the basis of their 1H NMR, 13C NMR, APT, COSY, TOCSY, ROESY, HSQC, and HMBC spectra. In accordance with the earlier reports [18] on structures of saponins from A. gypsophiloides, the aglycons of compound 1 and 2
were supposed to comprise quillaic acid (16-α-hydroxygypsogenin) and
gypsogenin, respectively. This assumption was in good agreement with the
detection of characteristic signals for six methyl groups in the 1H (Table 1) and 13C NMR (Table 2) spectra of 1 and 2. Furthermore, the presence of these aglycons was unambiguously confirmed by the good agreement between 13C NMR shifts of aglycon moieties of 1 and 2 and signals of aglycons for described bidesmosides comprising quillaic acid [21] and gypsogenin [21].| Comp. | C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 | C-8 | C-9 | C-10 | C-11 | C-12 | C-13 | C-14 | C-15 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H-1 | H-2 | H-3 | H-5 | H-6 | H-7 | H-9 | H-11 | H-12 | H-15 | ||||||
| 1 | 38.3 | 25.2 | 85.3 | 55.9 | 48.3 | 20.7 | 32.8 | 40.4 | 47.0 | 36.3 | 23.9 | 122.7 | 144.1 | 42.2 | 35.9 |
| 1.53 | 2.28 | 4.06 | 1.37 | 1.40 | 1.62 | 1.75 | 1.91 | 5.37 | 2.04 | ||||||
| 0.91 | 1.97 | 1.01 | 1.49 | 1.86 | 1.89 | ||||||||||
| 2 | 38.2 | 25.1 | 85.4 | 56.0 | 48.2 | 20.8 | 32.6 | 40.2 | 47.8 | 36.3 | 23.8 | 122.8 | 144.1 | 42.5 | 28.7 |
| 1.51 | 2.29 | 4.10 | 1.43 | 1.43 | 1.62 | 1.66 | 1.87 | 5.37 | 1.81 | ||||||
| 0.94 | 1.97 | 1.08 | 1.48 | 1.82 | 1.42 | ||||||||||
| Comp. | C-16 | C-17 | C-18 | C-19 | C-20 | C-21 | C-22 | C-23 | C-24 | C-25 | C-26 | C-27 | C-28 | C-29 | C-30 |
| H-16 | H-18 | H-19 | H-21 | H-22 | H-23 | H-24 | H-25 | H-26 | H-27 | H-29 | H-30 | ||||
| 1 | 73.9 | 47.9 | 41.6 | 47.4 | 29.3 | 35.8 | 31.5 | 211.6 | 10.7 | 16.0 | 17.6 | 27.3 | 177.1 | 33.1 | 24.6 |
| 5.01 | 3.27 | 2.57 | 2.19 | 2.28 | 9.71 | 1.43 | 0.88 | 0.96 | 1.68 | 0.94 | 0.96 | ||||
| 1.24 | 1.26 | 2.04 | |||||||||||||
| 2 | 23.3 | 47.9 | 42.1 | 46.4 | 30.8 | 33.9 | 32.4 | 211.5 | 10.7 | 15.8 | 17.5 | 26.1 | 176.4 | 33.2 | 23.7 |
| 2.05 | 2.99 | 1.68 | 1.25 | 1.82 | 9.63 | 1.43 | 0.85 | 0.92 | 1.24 | 0.93 | 0.85 | ||||
| 1.75 | 1.17 | 1.14 | 1.66 | ||||||||||||
| a1H NMR chemical shifts are italicized. | |||||||||||||||
| Units, atoms | 1 | 2 | |||
|---|---|---|---|---|---|
| δC | δH (J) | δC | δH (J) | ||
| →2,3)-GlcA (a) | |||||
| 1 | 103.4 | 4.83, d (7.8) | 103.4 | 4.82, d (7.3) | |
| 2 | 77.7 | 4.26 | 77.7 | 4.27 | |
| 3 | 85.0 | 4.30 | 85.0 | 4.31 | |
| 4 | 71.6 | 4.16 | 71.6 | 4.17 | |
| 5 | 77.7 | 4.26 | 77.7 | 4.27 | |
| 6 | 175.2 | 175.2 | |||
| Gal (b) | |||||
| 1 | 103.2 | 5.33, d (7.7) | 103.2 | 5.33, d (7.5) | |
| 2 | 72.8 | 4.14 | 72.8 | 4.14 | |
| 3 | 74.4 | 4.09 | 74.5 | 4.08 | |
| 4 | 70.3 | 4.31 | 70.3 | 4.31 | |
| 5 | 76.5 | 3.97 | 76.5 | 3.97 | |
| 6(a, b) | 62.2 | 4.33, 4.17 | 62.1 | 4.35, 4.17 | |
| Ara (c) | |||||
| 1 | 104.0 | 5.16, d (7.5) | 104.0 | 5.17, d (7.5) | |
| 2 | 72.4 | 4.23 | 72.4 | 4.23 | |
| 3 | 73.7 | 4.12 | 73.8 | 4.12 | |
| 4 | 69.3 | 4.28 | 69.4 | 4.28 | |
| 5(a, b) | 67.2 | 4.34, 3.95 | 67.2 | 4.34, 3.95 | |
| →2,4)-Fuc (d) | |||||
| 1 | 94.4 | 5.78, d (8.1) | 94.5 | 5.80, d (8.1) | |
| 2 | 74.6 | 4.43 | 75.1 | 4.41 | |
| 3 | 76.3 | 4.20 | 76.0 | 4.19 | |
| 4 | 83.2 | 4.12 | 83.0 | 4.12 | |
| 5 | 71.9 | 4.03 | 71.8 | 4.02 | |
| 6 | 17.1 | 1.52 | 17.1 | 1.52 | |
| Qui (e) | |||||
| 1 | 105.6 | 4.92, d (7.8) | 105.6 | 4.92, d (7.8) | |
| 2 | 75.6 | 3.81 | 75.6 | 3.80 | |
| 3 | 77.0 | 3.99 | 77.1 | 4.00 | |
| 4 | 76.1 | 3.53 | 76.1 | 3.53 | |
| 5 | 72.9 | 3.69 | 72.9 | 3.70 | |
| 6 | 18.2 | 1.51 | 18.2 | 1.51 | |
| →4)-Rha (f) | |||||
| 1 | 101.2 | 6.01 s (<1) | 101.2 | 5.97 s (<1) | |
| 2 | 71.1 | 4.62 | 71.1 | 4.62 | |
| 3 | 71.8 | 4.41 | 71.8 | 4.43 | |
| 4 | 83.7 | 4.15 | 83.7 | 4.18 | |
| 5 | 68.3 | 4.26 | 68.7 | 4.28 | |
| 6 | 18.3 | 1.65 | 18.4 | 1.68 | |
| →3)-Xyl (g) | |||||
| 1 | 106.1 | 5.06, d (8.5) | 105.9 | 5.09, d (7.7) | |
| 2 | 74.7 | 3.92 | 74.7 | 3.91 | |
| 3 | 86.5 | 4.02 | 86.4 | 4.01 | |
| 4 | 68.8 | 3.99 | 68.8 | 4.00 | |
| 5(a, b) | 66.2 | 4.18, 3.58 | 66.2 | 4.18, 3.58 | |
| Xyl (h) | |||||
| 1 | 104.9 | 5.03, d (8.8) | 104.9 | 5.04, d (7.6) | |
| 2 | 74.7 | 3.92 | 74.7 | 3.91 | |
| 3 | 77.0 | 4.01 | 77.1 | 4.01 | |
| 4 | 70.2 | 4.09 | 70.2 | 4.09 | |
| 5(a, b) | 66.5 | 4.30, 3.69 | 66.5 | 4.30, 3.68 | |
![[1860-5397-8-87-2]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-8-87-2.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 2:
Part of a 2D ROESY spectrum of compound 1. The corresponding parts of the 1H
NMR spectrum are shown along the axes. Arabic numerals refer to atoms
in sugar residues denoted by letters, as shown for compounds 1 and 2. Slashes are used for the designation of inter-residual interactions.
![[1860-5397-8-87-3]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-8-87-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3:
Key ROESY (dashed line) correlations for compound 1.
![[1860-5397-8-87-4]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-8-87-4.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 4:
Part of the HMBC spectrum of compound 1. 1H and 13C
NMR spectra are shown along the horizontal and vertical axes,
respectively. Arabic numerals before a slash refer to protons and after a
slash refer to carbons in sugar residues denoted by letters, as shown
for compounds 1 and 2.
Characteristic chemical shifts in the 13C NMR spectrum of 2 (δC 85.4 ppm for C-3 and δC
176.4 ppm for C-28 of the aglycon) evidence the bidesmosidic nature of
the genin, which is glycosydated at C-3 and esterified to an
oligosaccharide. The structures of both the trisaccharide and
pentasaccharide fragments of compound 2 are similar to those established for compound 1. Thus the structure of 2 was elucidated as gypsogenin 28-O-β-D-xylopyranosyl-(1→3)-β-D-xylopyranosyl-(1→3)-α-L-rhamnopyranosyl-(1→2)-[6-deoxy-β-D-glucopyranosyl-(1→4)]-β-D-fucopyranosyl ester 3-O-α-L-arabinopyranosyl-(1→3)-[β-D-galactopyranosyl-(1→2)]-β-D-glucuronopyranoside.Triterpenoid saponins from the roots of Acanthophyllum gypsophiloides Regel
, , , , , , , , , , , , , , , , and Nikolay E. Nifantiev1
1Laboratory of Glycoconjugate Chemistry, N. D. Zelinsky
Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky
prospect 47, 119991 Moscow, Russian Federation
2Laboratory of NMR spectroscopy, N. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky prospect 47, 119991 Moscow, Russian Federation
3Institute of Immunology, Ministry of Health and Social Development of Russian Federation, Kashirskoe Chausseе, 24/2, 115478 Moscow, Russian Federation
4Laboratory of Pharmacological Researches N. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, Siberian Branch of the Russian Academy of Sciences, prospect Acad. Lavrent’eva, 9, 630090 Novosibirsk, Russian Federation
5Institute of Cytology and Genetics Siberian Branch of the Russian Academy of Sciences, 10 prospect Acad. Lavrent’eva, 630090 Novosibirsk, Russian Federation
6Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 8 prospect Acad. Lavrent’eva, 630090 Novosibirsk, Russian Federation
7G. K. Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, 142290 Pushchino, Moscow region, Russian Federation
2Laboratory of NMR spectroscopy, N. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky prospect 47, 119991 Moscow, Russian Federation
3Institute of Immunology, Ministry of Health and Social Development of Russian Federation, Kashirskoe Chausseе, 24/2, 115478 Moscow, Russian Federation
4Laboratory of Pharmacological Researches N. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, Siberian Branch of the Russian Academy of Sciences, prospect Acad. Lavrent’eva, 9, 630090 Novosibirsk, Russian Federation
5Institute of Cytology and Genetics Siberian Branch of the Russian Academy of Sciences, 10 prospect Acad. Lavrent’eva, 630090 Novosibirsk, Russian Federation
6Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 8 prospect Acad. Lavrent’eva, 630090 Novosibirsk, Russian Federation
7G. K. Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, 142290 Pushchino, Moscow region, Russian Federation
Corresponding author email
This article is part of the Thematic Series "Synthesis in the glycosciences II".
Guest Editor: T. K. Lindhorst
Beilstein J. Org. Chem. 2012, 8, 763–775.
http://www.beilstein-journals.org/bjoc/single/articleFullText.htm?publicId=1860-5397-8-87//////
 .
.



 .
.






 .
. .
.








 .
.