05/05/2017
The U.S. Food and Drug Administration today approved Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease.
May 5, 2017
Release
The U.S. Food and Drug Administration today approved Radicava (edaravone) to treat patients with amyotrophic lateral sclerosis (ALS), commonly referred to as Lou Gehrig’s disease.
“After learning about the use of edaravone to treat ALS in Japan, we rapidly engaged with the drug developer about filing a marketing application in the United States,” said Eric Bastings, M.D., deputy director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This is the first new treatment approved by the FDA for ALS in many years, and we are pleased that people with ALS will now have an additional option.”
ALS is a rare disease that attacks and kills the nerve cells that control voluntary muscles. Voluntary muscles produce movements such as chewing, walking, breathing and talking. The nerves lose the ability to activate specific muscles, which causes the muscles to become weak and leads to paralysis. ALS is progressive, meaning it gets worse over time. The Centers for Disease Control and Prevention estimates that approximately 12,000-15,000 Americans have ALS. Most people with ALS die from respiratory failure, usually within three to five years from when the symptoms first appear.
Radicava is an intravenous infusion given by a health care professional. It is administered with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
The efficacy of edaravone for the treatment of ALS was demonstrated in a six-month clinical trial conducted in Japan. In the trial, 137 participants were randomized to receive edaravone or placebo. At Week 24, individuals receiving edaravone declined less on a clinical assessment of daily functioning compared to those receiving a placebo.
The most common adverse reactions reported by clinical trial participants receiving edaravone were bruising (contusion) and gait disturbance.
Radicava is also associated with serious risks that require immediate medical care, such as hives, swelling, or shortness of breath, and allergic reactions to sodium bisulfite, an ingredient in the drug. Sodium bisulfite may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The FDA granted this drug orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Radicava to Mitsubishi Tanabe Pharma America, Inc,

1-Phenyl-3-methyl-5
3H-Pyrazol-3-one, 2
89-25-8 [RN]
эдаравон [Russian]
إيدارافون [Arabic]
依达拉奉 [Chinese]
ラジカット,
Edaravone (brand name ラジカット, Radicut) is a nootropic and neuroprotective agent used for the purpose of aiding neurological recovery following acute brain ischemia and subsequent cerebral infarction.[1] It acts as a potent antioxidant and strongly scavenges free radicals, protecting against oxidative stress and neuronal apoptosis.[2][3][4] It has been marketed solely in Japan by Mitsubishi Pharma since 2001.[1] It is also marketed in India by Edinburgh Pharmaceuticals by the brand name Arone.
On June 26, 2015, Mitsubishi Tanabe Pharma Corporation announced it has received approval to market Radicut for treatment of ALS in Japan. The phase III clinical trial began in 2011 in Japan. The company was awarded Orphan Drug Designation for Radicut by the FDA and EU in 2015. Radicut is an intravenous drug and administrated 14 days followed by 14 days drug holiday.
The biotech company Treeway is developing an oral formulation of edaravone (TW001) and is currently in clinical development. Treeway was awarded orphan drug designation for edaravone by the EMA in November 2014 and FDA in January 2015.
Edaravone has been shown to attenuate methamphetamine- and 6-OHDA-induced dopaminergic neurotoxicity in the striatum and substantia nigra, and does not affect methamphetamine-induced dopamine release or hyperthermia.[5][6] It has also been demonstrated to protect against MPTP-mediated dopaminergic neurotoxicity to the substantia nigra, though notably not to the striatum.[7][8][9]

Edaravone (CAS NO.: 89-25-8), with other name of 3-Methyl-1-phenyl-2-pyrazolin-5-one, could be produced through many synthetic methods.
Following is one of the synthesis routes: By direct cyclization of phenylhydrazine (I) with ethyl acetoacetate (II) in refluxing ethanol.
SYNTHESIS
Edaravone, chemical name: 3-methyl-1-phenyl-2-pyrazoline-5-one, of the formula: Formula: CiciHltlN2O, molecular weight: 174.20, the formula:

[0004] Edaravone is a brain-protecting agent (free radical scavenger). Clinical studies suggest that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction. In patients with acute cerebral infarction Edaravone suppressed reduce peri-infarct regional cerebral blood flow, so that the first concept of days after the onset of brain NAA glycerol content than the control group significantly increased. Preclinical studies suggest that rats after ischemia / reperfusion of ischemic intravenous edaravone, can prevent the progress of cerebral edema and cerebral infarction, and relieve the accompanying neurological symptoms, suppress delayed neuronal death. Mechanism studies suggest that edaravone can scavenge free radicals, inhibiting lipid peroxidation, thereby inhibiting brain cells, endothelial cells, oxidative damage nerve cells.
For the synthesis of edaravone reported some use of benzene and methyl ethyl ketone amide corpus obtained, but methyl ethyl ketone amide difficult to obtain and slow reaction, which now has basically been abandoned; some use benzene corpus and ethyl acetoacetate in ethanol (see US4857542A, Synthesis Example 1) or water (Dykhanov NN Ethyl and butyl acetoacetates, Med Prom SSSR, 1961,15 (1):. 42-45) refluxing the reaction of the reaction The resulting purity edaravone poor, and the yield is not high, only about 70%.
Edaravone, chemical name: 2,4_-dihydro-5-methyl-2-phenyl pyrazole -3H- - one, of the formula: CiciHltlN2O, molecular weight: 174.20, the formula:

Edaravone is a clear cerebral infarction harmful factors (free radicals), protection of new therapeutic agents for cerebral infarction nerve cells. Clinical studies have shown that N- acetyl aspartate (NAA) is a specific sign of the survival of nerve cells, dramatically reducing the initial content of cerebral infarction. When patients with acute cerebral infarction Edaravone, peri-infarct rCBF decrease has improved, and the first 28 days after the onset of brain NAA content was significantly higher than that in the control group glycerol. Mechanism studies suggest that edaravone can clear the brain is highly cytotoxic hydroxyl radicals, inhibiting the synthesis of lipids free radicals, which can suppress brain infarction after reperfusion edema, protecting brain from damage and improve nerve impairment symptoms, and the delayed neuronal death inhibition, to protect the brain.
The first is by phenylhydrazine and methyl ethyl ketone amide (National API process compilation, 1980.737-739) condensation reaction in water at 50 ° C, a yield of up to 97%, but the raw material ketone amide ( CH3C0CH2C0NH2) are not readily available. Formula I
Edaravone synthetic route for the reaction:

[0008] The second is to phenylhydrazine and ethyl acetoacetate in ethanol or water at reflux the reaction, sodium bisulfite as the preparation of the catalyst. From the perspective of the chemical reaction, acetyl ethyl ketone amide more than hydrazine reacted with benzene and ethyl acetoacetate more readily available, the price is cheaper, but lower reaction yield of about 70%. Formula 2 for the synthesis route Edaravone reaction formula:

PATENT

1 Edaravone Synthesis Example [0023] Example
[0024] (1) Weigh benzene hydrochloride corpus 13. 5g (94mmol), was added to IOOml water, stirred for 0.5 hours, sodium hydroxide was added an equimolar 3. 76g, stirred for 0.5 hours; [0025] ( 2) To the reaction solution was added dropwise ethyl acetoacetate 11. 7g (90mmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 15. 5g;
[0026] (3) The crude product was added 30ml volume ratio of 2: 1 isopropanol - water, 2g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 14 a white crystalline powder. 8g, yield 90%, mpU9 ° C, with a purity of 99.9% 0
2 Edaravone Synthesis Example [0027] Example
[0028] (1) Weigh 15g of benzene hydrochloride corpus (I (Mmmol), was added to 120ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 4. 16g, stirred for 0.5 hours;
[0029] (2) To the reaction solution was added dropwise 13g of ethyl acetoacetate (lOOmmol), the reaction exotherm, the reaction was heated to reflux for 2.5 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 16. 7g;
[0030] (3) The crude product was added 40ml volume ratio of 2: 1 isopropanol - water, 2. 5g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature to precipitate a white solid, as a white crystalline powder 16. lg, a yield of 88.9%, mpU8 ° C, with a purity of 99.9% 0
3 Edaravone Synthesis Example [0031] Example
[0032] (1) Weigh 22g of benzene hydrochloride corpus (152mm0l), was added to 200ml of water and stirred for 0.5 hours, sodium hydroxide was added an equimolar 6. 08g, stirred for 0.5 hours;
[0033] (2) To the reaction solution was added dropwise 19g of ethyl acetoacetate (146mm0l), the reaction exotherm, the reaction was heated to reflux for 3 hours, heating was stopped, cooled to room temperature with stirring, filtered and dried to give a pale yellow granular crude 24. Sg;
[0034] (3) The crude product was added 50ml volume ratio of 2: 1 isopropanol - water, 3g of activated carbon was added and refluxed for 1 hour, filtered hot, cooled to room temperature a white solid was precipitated to give 23 a white crystalline powder. 2g, a yield of 87. 8%, mpU8 ° C, with a purity of 99.9% 0
[0035] Comparative Example
[0036] The ethyl acetoacetate 65g (0. 5mol) and 180ml of anhydrous ethanol mixed, with stirring at 50 ° C was added dropwise benzyl corpus 54g (0. 5mol) and a solution consisting of 30ml absolute ethanol, dropwise at reflux for 2 Bi hours, ethanol was distilled off 60ml, cooled, suction filtered, washed crystals with cold absolute ethanol twice, and dried in vacuo to give pale yellow crystals 70g. Recrystallized twice from absolute ethanol to give pale yellowish white crystals 56g (yield 65%).
PATENT
Example 1: Preparation of phenylhydrazine edaravone.
[0024] a. Weigh 5.1g phenylhydrazine (47mmol), was added under stirring to water containing 45mL round-bottom flask, take appropriate concentrated hydrochloric acid solution was adjusted to pH 6.0 with PH meter.
[0025] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, was added 1.5g sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0026] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 85% yield, 99.2% purity 0
[0027] Example 2: Preparation of phenylhydrazine hydrochloride edaravone.
[0028] a. Weigh 6.8g phenylhydrazine hydrochloride (47mmol), was added under stirring to water containing 45mL round-bottomed flask, the pH of the solution adjusted to 6.0 with aqueous ammonia.
[0029] b. To the above solution was slowly added dropwise ethyl acetoacetate 5.85g (45mmol), the reaction exotherm, 1.25g was added sodium dithionite (Na2S2O6), heated to 105 ° C to room temperature until reflux After 3h, heating was stopped, and then stirred, cooling, filtration, and dried to give a pale yellow granular edaravone crude.
[0030] c. With anhydrous ethanol recrystallization, filtration, and dried to obtain a white crystalline powder that is refined edaravone, 84% yield, with a purity of 99.2%. [0031] Comparative Example:
Under the [0032] state of agitation will phenylhydrazine 10.2g (94mmol) added to a round bottom flask equipped with IOOmL water in an appropriate amount of concentrated hydrochloric acid was dubbed the volume ratio of 1: 1 aqueous hydrochloric acid, with a PH adjusting pH of the solution was measured 6.0. After weighing Ethylacetoacetate 11.7g (90mmol) added to the reaction mixture, the reaction was exothermic and cooling to room temperature, sodium bisulfite (NaHSO3), heated to 105 ° C under reflux for 3h, the hot solution Water was added into the beaker and mechanical stirring, cooling, filtration, and dried to give the yellow edaravone crude, 73% yield, with a purity of 99.1%.

CLIP



Edaravone:
IR (KBr) max/cm-1 : 3431, 3129, 1602, 1599, 1580;
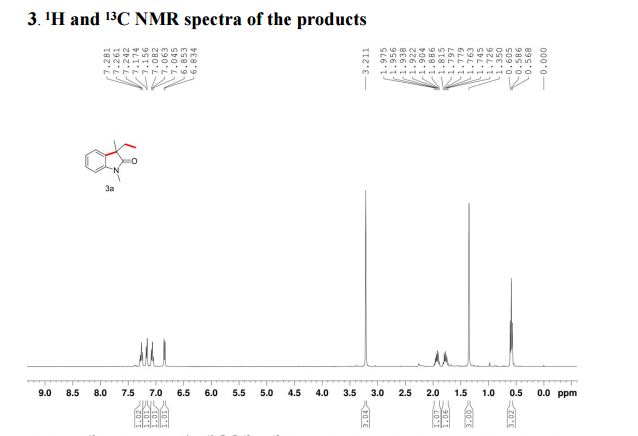
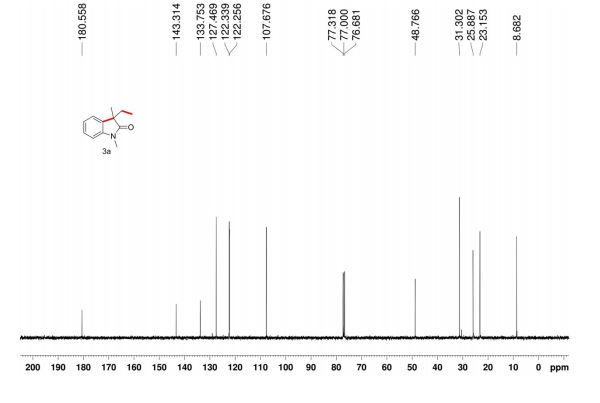
1 H NMR (300 MHz, CDCl3): δ 7.86 (d, J = 7.5 Hz, 2H, ArH), 7.40 (m, 2H, ArH), 7.18 (m, 1H, ArH), 3.41 (d, J =0.6 Hz, 2H, CH2), 2.19 (s, 3H, CH3);
13C NMR (75 MHz, CDCl3): 170.6, 156.4, 130.1, 128.8, 125.0, 118.9, 43.1, 17.0; 1 H NMR (300 MHz, DMSO-d6): δ 11.5 (bs, 1H, NH), 7.71 (m, 2H, ArH), 7.40 (m, 2H, ArH), 7.22 (m, 1H, ArH), 5.36 (s, 1H, CH), 2.12 (s, 3H, CH3);
13C NMR (75 MHz, DMSO-d6):171.7, 158.9, 148.7, 139.2, 138.6, 129.3,125.4, 124.8, 118.4, 43.5, 17.1, 14.2.
These values are in accordance with the previous published in literature1 .
In the carbon spectrum in DMSO presented in Figure SM 4.2.3.1.8 is evident the presence of the two major tautomeric structures of edaravone, signals are identified by different colours in both structures in the figure.
Also in the IR analysis of the solid material (Figure SM 4.2.3.1.9) is possible to see either the NH form (max/cm-1, 3129), the OH form (max/cm- 1 , 3431) and the C=O (max/cm-1, 1599) of the enol and keto tautomeric forms of edaravone.




CLIP
http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0103-50532008000600023
We have shown that the short reaction time, in combination with good yields can make microwave assisted reaction of hydrazines with β-ketoesters ideal for a rapid entry to pyrazolones. All the compounds synthesized are characterized by spectroscopic (1H NMR, IR and MS) data. While determination of tautomeric composition of compound 3 is quite challenging as eight possible tautomeric forms need to be considered, interestingly, two major tautomeric forms of compound 3a was observed in two different solvents. For example, it exists as 1,2-dihydro pyrazolone (T-1, Figure 2) in DMSO and 2,4-dihydro form (T-2, Figure 2) in chloroform as indicated by 1H NMR spectra (Figure 3). The olefinic proton of T-1 appeared at 5.36 δ whereas the methylene hydrogens appeared at 3.43 δ in case of T-2. Additionally, the NH proton of T-1 at 11.40 δ was not observed incase of T-2 confirmed the absence of NH in the 2,4-dihydro form. Existence of two major tautomeric forms was also observed in case compound 3b (see 1H NMR data in the experimental section). However, X-ray study on single crystal of 2-(4-chlorophenyl)-5-methyl-1,2-dihydro pyrazol-3-one (3i) indicates that 2-aryl pyrazol-3-ones e.g. 3a-b, 3e-f and 3i exist as 1,2-dihydro form in crystal state. 27 It is mention worthy that the aryl ring of all these 2-aryl pyrazol-3-ones remain twisted with respect to the pyrazole plane as indicated by the crystallographic data of 3i [the dihedral angle between the pyrazole and benzene ring planes was found to be 15.81 (11)º].27


5-Methyl-2-phenyl-1,2-dihydro pyrazol-3-one (3a)
mp 125-127 ºC (lit21 126-130 ºC);
IR (KBr) νmax/cm-1: 3127, 1597, 1525, 1498, 1454;
1H NMR (400 MHz, DMSO-d6) δ 11.40 (bs, 1H), 7.71-7.69 (m, 2H), 7.42-7.38 (m, 2H), 7.21-7.18 (m, 1H), 5.36 (s, 1H), 2.10 (s, 3H);
13C NMR (50 MHz, DMSO-d6) δ 170.6, 156.2, 138.1, 128.8 (2C), 124.9, 118.9 (2C), 43.1, 16.9;
Mass (CI, m/z) 175 (M+1, 100).
1H NMR (400 MHz, CDCl3)δ 7.85 (d, J 8.3 Hz, 2H), 7.40-7.37 (m, 2H), 7.24-7.18 (m, 1H), 3.43 (s, 2H), 2.20 (s, 3H).
21. Makhija, M. T.; Kasliwal, R. T.; Kulkarni, V. M.; Neamati, N.; Bioorg. Med. Chem. 2004, 12, 2317. [ Links ]
| CN101830852A | Mar 22, 2010 | Sep 15, 2010 | 海南美兰史克制药有限公司 | Edaravone compound synthesized by new method |
| CN102060771A | Nov 18, 2009 | May 18, 2011 | 南京长澳制药有限公司 | Edaravone crystal form and preparation method thereof |
| CN102180834A | Mar 24, 2011 | Sep 14, 2011 | 江苏正大丰海制药有限公司 | Preparation method for edaravone |
References
- ^ Jump up to:a b Doherty, Annette M. (2002). Annual Reports in Medicinal Chemistry, Volume 37 (Annual Reports in Medicinal Chemistry). Boston: Academic Press. ISBN 0-12-040537-7.
- Jump up^ Watanabe T, Tanaka M, Watanabe K, Takamatsu Y, Tobe A (March 2004). "[Research and development of the free radical scavenger edaravone as a neuroprotectant]". Yakugaku Zasshi (in Japanese). 124 (3): 99–111. doi:10.1248/yakushi.124.99. PMID 15049127.
- Jump up^ Higashi Y, Jitsuiki D, Chayama K, Yoshizumi M (January 2006). "Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases". Recent Patents on Cardiovascular Drug Discovery. 1 (1): 85–93. doi:10.2174/157489006775244191. PMID 18221078.
- Jump up^ Yoshida H, Yanai H, Namiki Y, Fukatsu-Sasaki K, Furutani N, Tada N (2006). "Neuroprotective effects of edaravone: a novel free radical scavenger in cerebrovascular injury". CNS Drug Reviews. 12 (1): 9–20. doi:10.1111/j.1527-3458.2006.00009.x. PMID 16834755.
- Jump up^ Yuan WJ, Yasuhara T, Shingo T, et al. (2008). "Neuroprotective effects of edaravone-administration on 6-OHDA-treated dopaminergic neurons". BMC Neuroscience. 9: 75. doi:10.1186/1471-2202-9-75. PMC 2533664
 . PMID 18671880.
. PMID 18671880. - Jump up^ Kawasaki T, Ishihara K, Ago Y, et al. (August 2006). "Protective effect of the radical scavenger edaravone against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum". European Journal of Pharmacology. 542 (1-3): 92–9. doi:10.1016/j.ejphar.2006.05.012. PMID 16784740.
- Jump up^ Kawasaki T, Ishihara K, Ago Y, Baba A, Matsuda T (July 2007). "Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a radical scavenger, prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in the substantia nigra but not the striatum". The Journal of Pharmacology and Experimental Therapeutics. 322 (1): 274–81. doi:10.1124/jpet.106.119206. PMID 17429058.
- Jump up^ Yokoyama H, Takagi S, Watanabe Y, Kato H, Araki T (June 2008). "Role of reactive nitrogen and reactive oxygen species against MPTP neurotoxicity in mice". Journal of Neural Transmission (Vienna, Austria : 1996). 115 (6): 831–42. doi:10.1007/s00702-008-0019-6. PMID 18235988.
- Jump up^ Yokoyama H, Yano R, Aoki E, Kato H, Araki T (September 2008). "Comparative pharmacological study of free radical scavenger, nitric oxide synthase inhibitor, nitric oxide synthase activator and cyclooxygenase inhibitor against MPTP neurotoxicity in mice". Metabolic Brain Disease. 23 (3): 335–49. doi:10.1007/s11011-008-9096-3. PMID 18648914.
External links
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Radicut |
| Routes of administration | Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| Synonyms | MCI-186 |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.001.719 |
| Chemical and physical data | |
| Formula | C10H10N2O |
| Molar mass | 174.20 g/mol |
| 3D model (Jmol) | |
////////// Radicava, edaravone, fda 2017, Lou Gehrig’s disease, amyotrophic lateral sclerosis, Mitsubishi Tanabe, orphan drug designation, 89-25-8, эдаравон, إيدارافون , 依达拉奉 ,ラジカット,
O=C1CC(=NN1c1ccccc1)C
O=C1CC(=NN1c1ccccc1)C





























 Open Access
Open Access