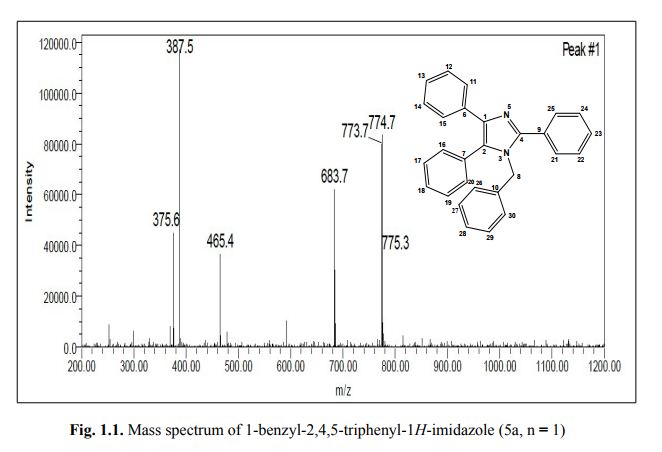
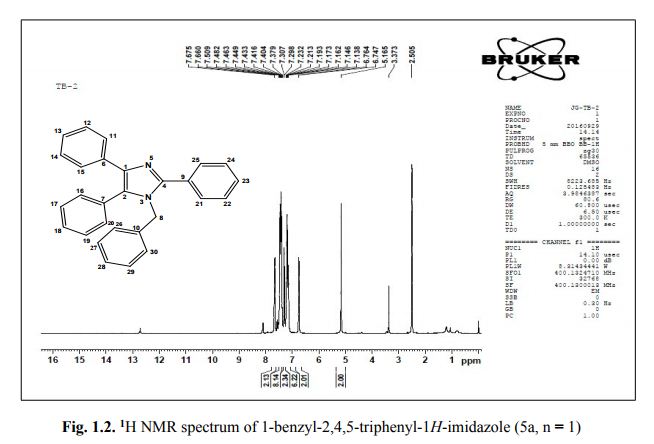
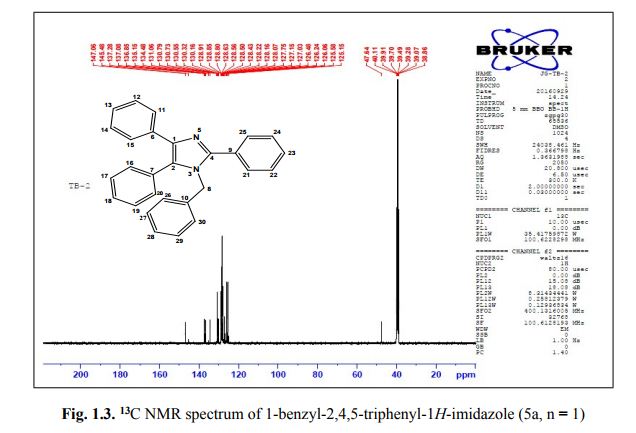
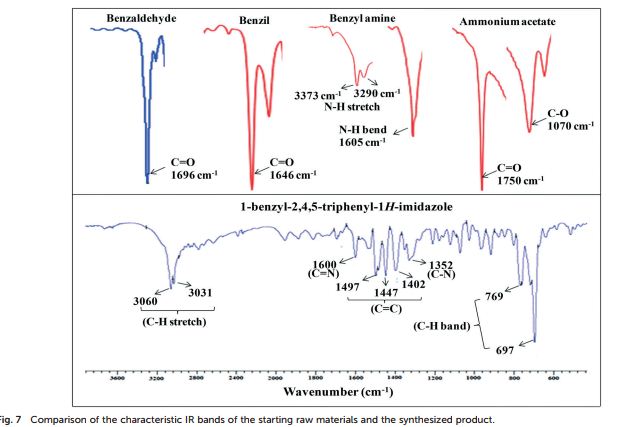
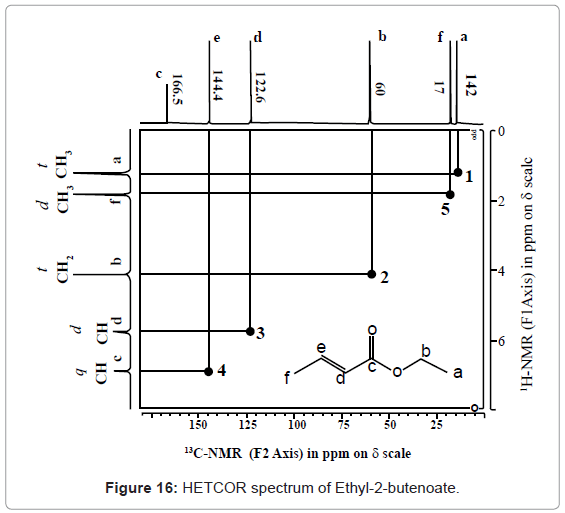
1-benzyl-2, 4, 5-triphenyl-1H-imidazole
. 1-Benzyl-2,4,5-triphenyl-1H-imidazole (5a, n = 1).
Off-white solid; m.p.: 160–162 °C;
anal. calcd. for C28H22N2: C, 87.01, H, 5.74, N, 7.25%. Found: C, 87.13, H, 5.70, N, 7.19%;
UV (λmax, ethanol) = 280 nm;
FT-IR (KBr, cm−1 ): 3060 (C–H stretch), 3031, 1600 (CN), 1497, 1483, 1447 (CC), 1352 (C–N stretch), 769, 697 (C–H band);
1 H NMR (400 MHz, DMSO): 5.16 (s, 2H, CH2), 6.74–7.67 (m, 20H, Ar–H) ppm;
13C NMR (100 MHz, DMSO): 47.6 (CH2, C8), 125.1 (CHarom, C28), 126.0 (CHarom, C26), 126.2 (CHarom, C30), 126.4 (CHarom, C11), 127.0 (CHarom, C15), 127.1 (CHarom, C16), 127.7 (CHarom, C20), 128.0 (CHarom, C21), 128.1 (CHarom, C25), 128.4 (CHarom, C13), 128.5 (CHarom, C18), 128.6 (CHarom, C27), 128.8 (C1), 128.8 (CHarom, C12), 128.9 (CHarom, C14), 130.1 (CHarom, C17), 130.3 (CHarom, C19), 130.5 (CHarom, C22), 130.7 (CHarom, C24), 131.0 (CHarom, C29), 134.4 (CHarom, C9), 135.1 (CHarom, C23), 136.8 (CHarom, C7), 137.0 (CHarom, C10), 137.2 (CHarom, C6), 145.4 (C2), 147.0 (C4) ppm;
MS: m/z = 387.5 (M + H)+
An efficient green protocol for the synthesis of tetra-substituted imidazoles catalyzed by zeolite BEA: effect of surface acidity and polarity of zeolite
S V National Institute of Technology, India
Jenifer J Gabla has obtained her MSc degree in Organic Chemistry, in the year 2013 from Uka Tarsadia University, Bardoli, Gujarat. She is currently pursuing her PhD in the area of solid acid catalyzed multicomponent reactions for the synthesis of biologically active drug molecules, at Applied Chemistry Department (ACD), S. V. National Institute of Technology (SVNIT) under the guidance of Kalpana Maheria, Assistant Professor & Head, ACD, SVNIT, Surat, Gujarat, India. Her research focuses on development of novel zeolite based catalytic materials and exploring their utility in the green process development for the synthesis of medicinal compounds. She has presented her research in several national and international conferences.
*Corresponding authors
aApplied Chemistry Department, S. V. National Institute of Technology, Ichchhanath, Surat – 395007, India
E-mail:kcmaheria@gmail.com
bMandvi Science College, Mandvi – 394160, Surat, India
Abstract
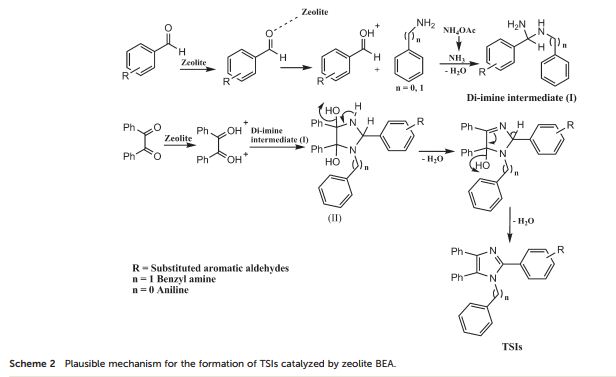
In the present study, the catalytic activity of various medium (H-ZSM-5) and large pore (H-BEA, H-Y, H-MOR) zeolites were studied as solid acid catalysts. The zeolite H-BEA is found to be an efficient catalyst for the synthesis of 1-benzyl-2,4,5-triphenyl-1H-imidazoles through one-pot, 4-component reaction (4-CR) between benzil, NH4OAc, substituted aromatic aldehydes and benzyl amine. The hydrophobicity, Si/Al ratio and acidic properties of zeolite BEA were well improved by controlled dealumination. The synthesized materials were characterized by various characterization techniques such as XRD, ICP-OES, BET, NH3-TPD, FT-IR, pyridine FT-IR, 27Al and 1H MAS NMR. It has been observed that the dealumination of the parent zeolite H-BEA (12) results in the enhanced strength of Brønsted acidity up to a certain Si/Al ratio which is attributed to the inductive effect of Lewis acidic EFAl species, leading to the higher activity of the zeolite BEA (15) catalyst towards the synthesis of 1-benzyl-2,4,5-triphenyl-1H-imidazoles under thermal solvent-free conditions with good to excellent yields. Using the present catalytic synthetic protocol, diverse tetra-substituted imidazoles, which are among the significant biologically active scaffolds, were synthesized in high yield within a shorter reaction time. The effect of polarity, surface acidity and extra framework Al species of the catalysts has been well demonstrated by means of pyridine FT-IR, and 27Al and 1H MAS NMR. The solvent-free synthetic protocol makes the process environmentally benign and economically viable.





S. V. National Institute of Technology, Ichchhanath, Surat
Mandvi Science College, Mandvi – 394160, Surat, India
////////
DISCLAIMER
“ORGANIC SPECTROSCOPY INT” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This is a compilation for educational purposes only. P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent