Copper-catalyzed pyrrole synthesis from 3,6-dihydro-1,2-oxazines
Abstract
Highly-functionalized pyrroles could be effectively synthesized from 3,6-dihydro-1,2-oxazines using a heterogeneous copper on carbon (Cu/C) under neat heating conditions. Furthermore, the in situ formation of 3,6-dihydro-1,2-oxazines via the hetero Diels–Alder reaction between nitroso dienophiles and 1,3-dienes and the following Cu/C-catalyzed pyrrole synthesis also provided the corresponding pyrrole derivatives in a one-pot manner.

Brown solid; M. p. 107–108 o C;
IR (ATR) cm-1 : 3064, 2923, 2851, 1687, 1596, 1562, 1541, 1498, 1488, 1459, 1451, 1422, 1390, 1343, 1319, 1256, 1187, 1098, 1073, 1053, 1037, 1009;
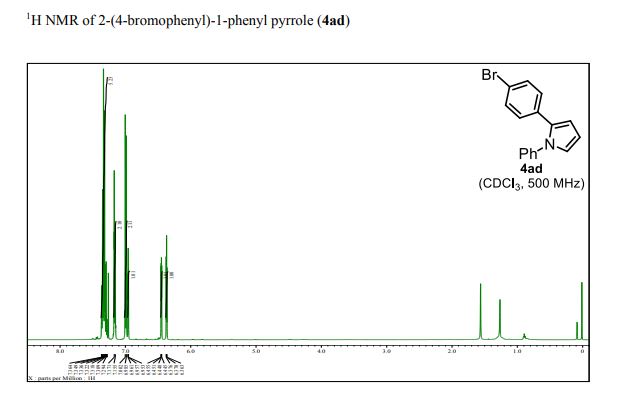
1 H NMR (500 MHz, CDCl3): δ 7.37–7.28 (m, 5H), 7.17 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 6.95 (dd, J = 2.0, 3.0 Hz, 1H), 6.45 (dd, J = 2.0, 3.0 Hz, 1H), 6.37 (dd, J = 3.0, 3.0 Hz, 1H);
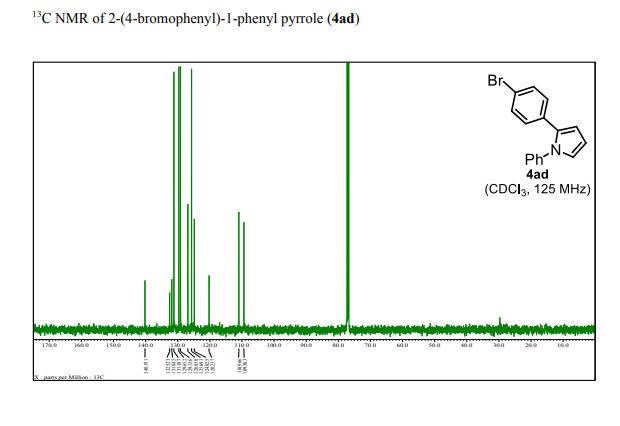
13C NMR (125 MHz, CDCl3): δ 140.19, 132.52, 131.85, 131.19, 129.65, 129.14, 126.84, 125.69, 124.83, 120.24, 110.97, 109.38;
ESI-HRMS m/z: 298.0231([M+H+ ]); Calcd for C16H13NBr: 298.0226.


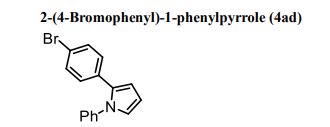
Brown solid; M. p. 107–108 o C;
IR (ATR) cm-1 : 3064, 2923, 2851, 1687, 1596, 1562, 1541, 1498, 1488, 1459, 1451, 1422, 1390, 1343, 1319, 1256, 1187, 1098, 1073, 1053, 1037, 1009;
1 H NMR (500 MHz, CDCl3): δ 7.37–7.28 (m, 5H), 7.17 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 6.95 (dd, J = 2.0, 3.0 Hz, 1H), 6.45 (dd, J = 2.0, 3.0 Hz, 1H), 6.37 (dd, J = 3.0, 3.0 Hz, 1H);
13C NMR (125 MHz, CDCl3): δ 140.19, 132.52, 131.85, 131.19, 129.65, 129.14, 126.84, 125.69, 124.83, 120.24, 110.97, 109.38;
ESI-HRMS m/z: 298.0231([M+H+ ]); Calcd for C16H13NBr: 298.0226.
//////////3,6-dihydro-1,2-oxazines