137-45-1 cas
- 3-Pyrazolin-5-one (8CI)
- Pyrazol-3(or 5)-ol (6CI,7CI)
- 1,2-Dihydro-3H-pyrazol-3-one
- 1,2-Dihydropyrazol-3-one
- 1H-Pyrazol-3-ol
- 1H-Pyrazol-5-ol
- 3-Hydroxypyrazole
- 3-Pyrazoline-5-one
- 3-Pyrazolone
- 4-Pyrazolin-3-one
- NSC 520837
- Pyrazol-3-ol
- Pyrazol-5-ol
Compound 1: Under stirring, to a solution of 5.81 g (50 mmol) of methyl (2E)-3-methoxyacrylate in methanol (5 mL) was hydrazine hydrate (2.75 g, 55 mmol) added and the mixture was refluxed for 1h. Evaporation under reduced pressure to dryness gave 4.13 g (98%) of a slightly yellowish powder, pure according to 1H NMR spectroscopy.
Melting point: 160–162 °C, crystal modifications starting at ~140 °C, (lit. [12] 162–164 °C).
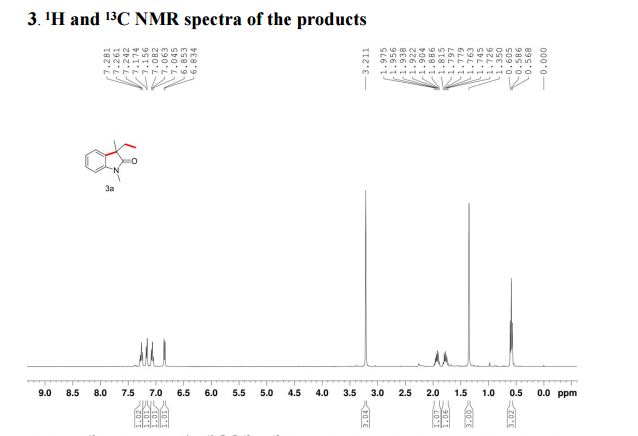
1H-NMR (300 MHz, DMSO-d6, 28 °C, numbering for 1H-pyrazol-3-ol = form D) [13]: δ= 9.82 (br s, 2H, XH); 7.33 (d, 3 J(H5,H4)= 2.3 Hz, 1H, H5); 5.43 (d, 3 J(H4,H5)= 2.3 Hz, 1H, H4).
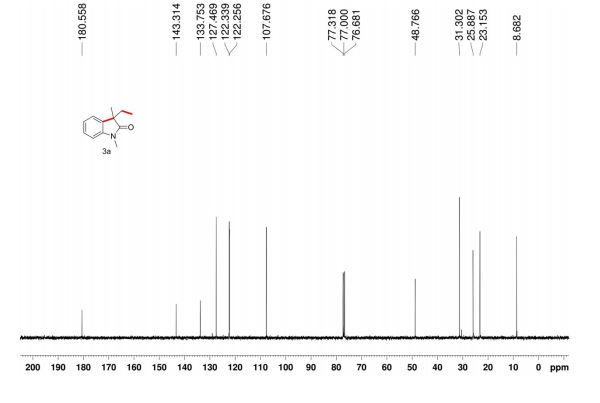
13C-NMR (75 MHz, DMSO-d6, 28 °C, numbering for 1H-pyrazol-3-ol = form D) [13]: δ= 161.0 (C3, 2 J(C3,H4)= 3.4 Hz, 3 J(C3,H5)= 9.2 Hz); 130.1 (C5, 1 J = 184.0 Hz, 2 J(C5,H4)= 8.2 Hz); 89.3 (C4, 1 J = 175.6 Hz, 2 J(C4,H5)= 8.7 Hz).
15N-NMR (50 MHz, DMSO-d6, 294 K) [14]: δ= –126.5; –192.0.
MS (m/z, %) [15]: 84 (M+ , 100); 55 (24).
Elemental Analysis: Calculated for C3H4N2O (84.08): C, 42.86%; H, 4.80%; N, 33.32%. Found: C, 42.75%; H, 4.65%; N, 33.15%.
References and /Notes:
1. J. Elguero, In 'Comprehensive Heterocyclic Chemistry: Pyrazoles and their Benzo Derivatives', Vol. 5; A. R. Katritzky and C. W. Rees, Eds., Pergamon Press, Oxford, 1984, 167–303.
2. Stanovnik, B.; Svete, J. Product class 1: Pyrazoles. Science of Synthesis 2002, 12, 15–225.
3. Eller, G. A.; Holzer, W. Heterocycles 2004, 63, 2537–2555.
4. Becker, W.; Eller, G. A.; Holzer, W. Synthesis 2005, 2583–2589.
5. Testa, E.; Fontanella, L. Farmaco 1971, 26, 1017–35.
6. Dorn, H.; Zubek, A. J. Prakt. Chem. 1971, 313, 1118–24.
7. Maywald, V.; Steinmetz, A.; Rack, M.; Gotz, N.; Gotz, R.; Henkelmann, J.; Becker, H.; Aiscar Bayeto, PCT Int. Appl. WO 0031042 A2 2000 (Chem. Abstr., 2000, 133, 4655).
8. Holzer, W.; Hallak, L. Heterocycles 2004, 63, 1311–1334, and references cited therein.
9. Cizmarik, J.; Lycka, A. Pharmazie 1988, 43, 794–795.
10. Holzer, W.; Kautsch, C.; Laggner, C.; Claramunt, R. M.; Perez-Torralba, M.; Alkorta, I.; Elguero, J. Molbank 2004 http://www.mdpi.org/molbank/molbank2006/m464.htm 2 von 3 24.02.2009 12:54 Tetrahedron 2004, 60, 6791–6805.
11. Sackus, A.; Holzer, W. manuscript in preparation.
12. Lingens, F.; Schneider-Bernloehr, H. Liebigs Ann. Chem. 1965, 686, 134–144.
13. The spectrum was obtained on a Varian UnityPlus 300 spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C). The center of the solvent signal was used as an internal standard which was related to TMS with δ 2.49 ppm (1H NMR) and δ 39.5 ppm (13C NMR).
14. The spectrum was obtained on a Bruker Avance 500 spectrometer and was referenced against neat, external nitromethane (coaxial capillary). The signals were not unequivocally assigned to the N atoms. 15. The spectrum was obtained on a Shimadzu QP 1000 instrument (EI, 70eV).
Molbank 2006, M464 http://www.mdpi.net/molbank/ A one-step synthesis of pyrazolone Gernot A. Eller* and Wolfgang Holzer Department of Drug Synthesis, University of Vienna, Althanstrasse 14, 1090 Vienna, Austria Phone: +43-1-4277-55634, e-mail: gernot.eller@univie.ac.at *Author to whom correspondence should be addressed
file:///C:/Users/91200291/Downloads/molbank-2006-M464.pdf
NMR IS EASY EVEN MOM CAN TEACH YOU
Patent
Synthesis of lH-pyrazol-3-ol
[0223] To a 100 mL round-bottom flask was added methyl (2E)-3-methoxyprop-2-enoate (11.6 g, 99.90 mmol, 1.00 equiv) and methanol (10.0 mL), followed by the addition of hydrazine hydrate (7.8 mL) dropwise with stirring. The resulting solution was stirred for 90 min at 85°C, then concentrated under vacuum to afford crude lH-pyrazol-3-ol as a white solid.
/////////////