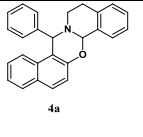
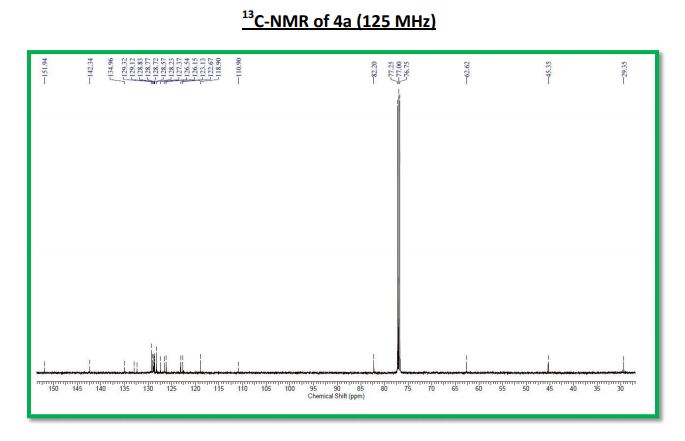
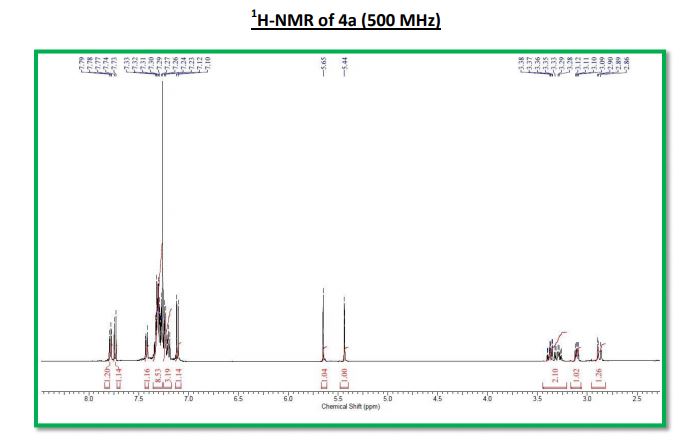
Cross dehydrogenative coupling of N-aryltetrahydroisoquinolines (sp3 C-H) with indoles (sp2 C-H) using a heterogeneous mesoporous manganese oxide catalyst
Green Chem., 2017, Advance Article
DOI: 10.1039/C7GC01919J, Communication
DOI: 10.1039/C7GC01919J, Communication
B. Dutta, V. Sharma, N. Sassu, Y. Dang, C. Weerakkody, J. Macharia, R. Miao, A. R. Howell, S. L. Suib
We disclose a novel, heterogeneous catalytic approach for selective coupling of C1 of N-aryltetrahydroisoquinolines with C3 of indoles in the presence of mesoporous manganese oxides.
We disclose a novel, heterogeneous catalytic approach for selective coupling of C1 of N-aryltetrahydroisoquinolines with C3 of indoles in the presence of mesoporous manganese oxides.
Cross dehydrogenative coupling of N-aryltetrahydroisoquinolines (sp3 C–H) with indoles (sp2 C–H) using a heterogeneous mesoporous manganese oxide catalyst


Biswanath Dutta
Ph.D Candidate
Chemistry
B.Sc. Chemistry, University of Calcutta, India, 2011
M.S. Chemistry, IIT Bombay, India, 2013
Group Member Since 2013
Research Area: Material synthesis, Catalysis

Steven Suib
Professor
| steven.suib@uconn.edu | |
| Phone | (860) 486-2797 |
| Fax | (860) 486-2981 |
| Mailing Address | University of Connecticut Department of Chemistry 55 N. Eagleville Rd Storrs, CT 06269 |
| Office Location | CHEM A-313 |
Abstract
We disclose a novel, heterogeneous catalytic approach for selective coupling of C1 of N-aryltetrahydroisoquinolines with C3 of indoles in the presence of mesoporous manganese oxides. Our work involves a detailed mechanistic investigation of the reaction on the catalyst surface, backed by DFT computational studies, to understand the superior catalytic activity of manganese oxides.



////////////////////