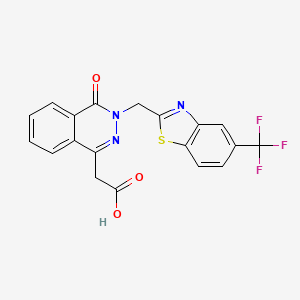
You will react cyclopentadiene with maleic anhydride to form the Diels-Alder product below. This Diels-Alder reaction produces almost solely the endo isomer upon reaction at ambient temperature.

The preference for endo–stereochemistry is “observed” in most Diels-Alder reactions. The fact that the more hindered endo product is formed puzzled scientists until Woodward, Hoffmann, and Fukui used molecular orbital theory to explain that overlap of the p orbitals on the substituents on the dienophile with p orbitals on the diene is favorable, helping to bring the two molecules together.
Hoffmann and Fukui shared the 1981 Nobel Prize in chemistry for their molecular orbital explanation of this and other organic reactions. In the illustration below, notice the favorable overlap (matching light or dark lobes) of the diene and the substituent on the dienophile in the formation of the endo product:

Oftentimes, even though the endo product is formed initially, an exo isomer will be isolated from a Diels-Alder reaction. This occurs because the exo isomer, having less steric strain than the Endo , is more stable, and because the Diels-Alder reaction is often reversible under the reaction conditions. In a reversible reaction, the product is formed, reverts to starting material, and forms again many times before being isolated.
The more stable the product, the less likely it will be to revert to the starting material. The isolation of an exo product from a Diels-Alder reaction is an example of an important concept: thermodynamic vs kinetic control of product composition. The first formed product in a reaction is called the kinetic product. If the reaction is not reversible under the conditions used, the kinetic product will be isolated. However, if the first formed product is not the most stable product and the reaction is reversible under the conditions used, then the most stable product, called the thermodynamic product, will often be isolated.
The NMR spectrum of cis-5-norbornene-2,3-endo-dicarboxylic anhydride is given below:

Cis-Norbornene-5,6-endo-dicarboxylic anhydride
Cyclopentadiene was previously prepared through the cracking of dicyclopentadiene and kept under cold conditions. In a 25 mL Erlenmeyer flask, maleic anhydride (1.02 g, 10.4 mmol) and ethyl acetate (4.0 mL) were combined, swirled, and slightly heated until completely dissolved. To the mixture, ligroin (4 mL) was added and mixed thoroughly until dissolved. Finally, cyclopentadiene (1 mL, 11.9 mmol) was added to the mixture and mixed extensively. The reaction was cooled to room temperature and placed into an ice bath until crystallized. The crystals were isolated through filtration in a Hirsch funnel. The product had the following properties: 0.47 g (27.6% yield) mp: 163-164 °C (lit: 164 °C). 1H NMR (CDCl3, 300 MHz) δ: 6.30 (dd, J=1.8 Hz, 2H), 3.57 (dd, J=7.0 Hz, 2H), 3.45 (m, 2H), 1.78 (dt, J=9.0,1.8 Hz, 1H), 1.59 (m, 1H) ppm. 13C NMR (CDCl3, 75Hz) δ: 171.3, 135.5, 52.7, 47.1, 46.1 ppm. IR 2982 (m), 1840 (s), 1767 (s), 1089 (m) cm-1.
Reaction Mechanism The scheme below depicts the concerted mechanism of the Diels-Alder reaction of cyclopentadiene and maleic anhydride to formcis-Norbornene-5,6-endo-dicarboxylic anhydride.

Results and Discussion
When combining the reagents, a cloudy mixture was produced and problems arose in the attempt to completely dissolve the mixture. After heating for about 10 minutes and magnetically stirring, tiny solids still remained. The undissolved solids were removed form the hot solution by filtration and once they cooled, white crystals began to form. Regarding the specific reaction between cyclopentadiene and maleic anhydride, the endo isomer, the kinetic product, was formed because the experiment was directed under mild conditions. The exo isomer is the thermodynamic product because it is more stable.3
When combining the reagents, a cloudy mixture was produced and problems arose in the attempt to completely dissolve the mixture. After heating for about 10 minutes and magnetically stirring, tiny solids still remained. The undissolved solids were removed form the hot solution by filtration and once they cooled, white crystals began to form. Regarding the specific reaction between cyclopentadiene and maleic anhydride, the endo isomer, the kinetic product, was formed because the experiment was directed under mild conditions. The exo isomer is the thermodynamic product because it is more stable.3
A total of 0.47 g of the product was collected; a yield of 27.6%. The melting point was in the range of 163-164 °C which indicates the absence of impurities because the known melting point of the product is 164 °C.

The 1H NMR spectrum of the product revealed a peak in the alkene range at 6.30 ppm, H-2 and H-3 (Figure 1). In addition, it exhibited two peaks at 3.57 and 3.45 ppm because of the proximity of H-1, H-4, H-5, and H-6 to an electronegative atom, oxygen. Finally, two peaks at 1.78 and 1.59 ppm corresponded to the sp3 hydrogens, Hb and Ha, respectively. Impurities that appeared included ethyl acetate at 4.03, 2.03, and 1.31 ppm as well as acetone at 2.16 ppm.
Regarding the 13C NMR, a peak appeared at 171.3 ppm, accounting for the presence of two carbonyl functional groups, represented by C-7 and C-8 in Figure 1. The alkene carbons, C-2 and C-3, exhibited a peak at 135.5 ppm, while the sp3 carbons close to oxygen, C-5 and C-6, displayed a peak at 52.7 ppm. Finally, peaks at 46.1 and 47.1 ppm accounted for the sp3 carbons, C-1 and C-4, and C-9. Impurities of ethyl acetate appeared at 46.6, 25.8, and 21.0 ppm accompanied with acetone at 30.9 ppm.
The IR spectrum revealed a peak at 2982 cm-1 representing the C-H stretches. A peak at 1840 cm-1 accounted for the carbonyl functional group, while a peak at 1767 cm-1 accounted for the alkene bond. A peak at 1089 cm-1 represented the carbon-oxygen functional group.
In order to distinguish between the two possible isomers, properties such as melting point and spectroscopy data were analyzed. The exo product possessed a melting point in the range of 140-145 °C which is significantly lower than the endo product. The observed melting point in this experiment supported the production of the endo isomer.
The 1H NMR spectum exhibited a doublet of doublets at 3.57 ppm for the endo isomer. The exo isomer would possess a triplet around 3.50 ppm due to the difference in dihedral angle between the hydrogen molecules of H-1 and H-4, and H-5 and H-6 (Figure 1). A peak at 3.00 ppm would appear in the exo isomer spectra as opposed to a peak at 3.60 ppm as shown in the observed endo product.3 This is because of the interaction and coupling with the H-5 and H-6, as displayed in Figure 1.
The 1H NMR spectum exhibited a doublet of doublets at 3.57 ppm for the endo isomer. The exo isomer would possess a triplet around 3.50 ppm due to the difference in dihedral angle between the hydrogen molecules of H-1 and H-4, and H-5 and H-6 (Figure 1). A peak at 3.00 ppm would appear in the exo isomer spectra as opposed to a peak at 3.60 ppm as shown in the observed endo product.3 This is because of the interaction and coupling with the H-5 and H-6, as displayed in Figure 1.
Conclusion
Through the Diels-Alder reaction, 27.6% yield of cis-Norbornene-5,6-endo-dicarboxylic anhydride was produced. The distinction of the presence of the endo isomer was proven by analyzing physical properties of both possible isomers.
Through the Diels-Alder reaction, 27.6% yield of cis-Norbornene-5,6-endo-dicarboxylic anhydride was produced. The distinction of the presence of the endo isomer was proven by analyzing physical properties of both possible isomers.
Martin, J.; Hill, R.; Chem Rev, 1961, 61, 537-562.
2 Pavia, L; Lampman, G; Kriz, G; Engel, R. A Small Scale Approach to Organic Laboratory Techniques, 2011, 400-409.
3 Myers, K.; Rosark, J. Diels-Alder Synthesis, 2004, 259-265.
link
http://orgspectroscopyint.blogspot.in/2014/08/cis-norborene-56-endo-dicarboxylic.html



 amcrasto@gmail.com
amcrasto@gmail.com
link
http://orgspectroscopyint.blogspot.in/2014/08/cis-norborene-56-endo-dicarboxylic.html
ORGANIC SPECTROSCOPY INTERNATIONAL
Read all about Organic Spectroscopy onORGANIC SPECTROSCOPY INTERNATIONAL 
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS, BRAVESITES,GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO, YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN,FRIENDFEED, NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are 
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT,SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD,LINKEDIN OPD, WDT LINKEDIN, WDTI ZING
amcrasto@gmail.com