 .
.Terretrione D
Key COSY and HMBC correlations of 2.

Figure 1.
Structures of Compounds 1–6.
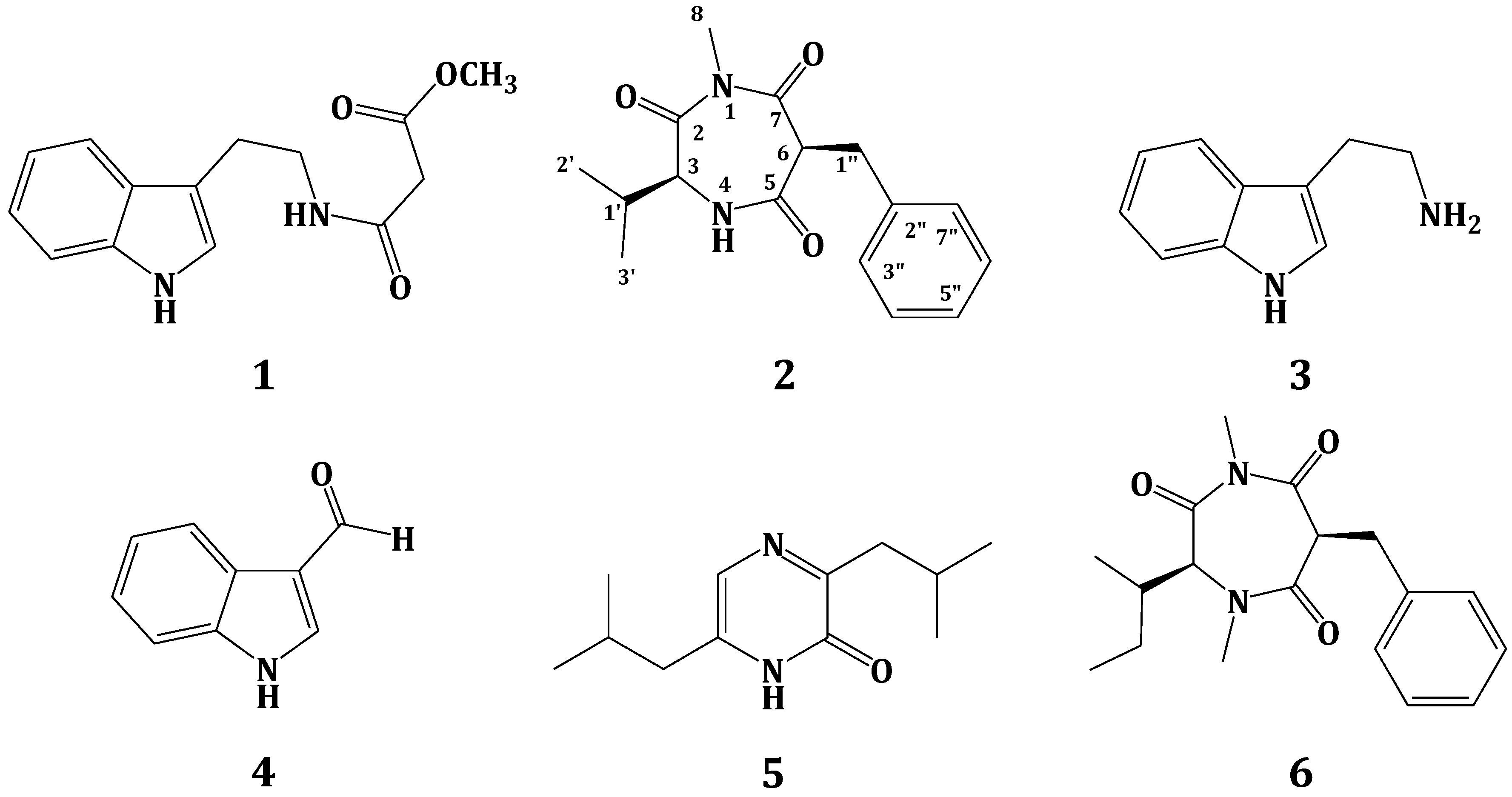
methyl-2-([2-(1H-indol-3-yl)ethyl]carbamoyl)acetate (1),
Terretrione D (2)
tryptamine (3),
indole-3-carbaldehyde (4),
3,6-diisobutylpyrazin-2(1H)-one (5) and
terretrione C (6),
| Position | δC, Type a | δH, m (J in Hz) | HMBC | NOESY |
|---|---|---|---|---|
| 2 | 173.0, C b | |||
| 3 | 60.1, CH | 4.18, d (4.8) | C-2, C-5, C-1′ | H-6, H3-8, H-1′ |
| 5 | 173.2, C | |||
| 6 | 56.4, CH | 4.68, dd (8.4, 6.6) | C-5, C-7, C-1″ | H-3, H3-8, H2-1″ |
| 7 | 173.1, C b | |||
| 8 | 22.9, CH3 | 1.90, s | C-2, C-7 | |
| 1′ | 32.2, CH | 2.07, m | C-2, C-3, C-2′, C-3′ | H-3 |
| 2′ | 19.8, CH3 | 0.81, d (7.2) | C-1′, C-3 | |
| 3′ | 18.2, CH3 | 0.78, d (7.2) | C-1′, C-3 | |
| 1″ | 39.0, CH2 | 3.11, dd (13.8, 6.6) 2.88, dd (13.8, 8.4) | C-2″, C-3″, C-6, C-7 | H-6 |
| 2″ | 138.6, C | |||
| 3″ | 130.2, CH | 7.23, m | C-2″, C-5″ | |
| 4″ | 129.4, CH | 7.24, m | C-2″ | |
| 5″ | 127.7, CH | 7.14, m | C-3″, C-7″ | |
| 6″ | 129.4, CH | 7.24, m | C-2″ | |
| 7″ | 130.2, CH | 7.23, m | C-1″, C-5″ |
a Multiplicities were deduced by DEPT and HSQC; b signals may be interchangeable.
compound 2 (Figure 1) showed a molecular formula C16H20N2O3, as deduced from the HRESIMS pseudomolecular ion peak at m/z 289.1555 [M + H]+, requiring eight degrees of unsaturation. The 13C NMR spectrum revealed signals for 16 carbons. Careful investigation of the 1H, 13C and 2D NMR spectra supported the presence of a valine residue in 2. This was evident from the NMR signals at δH/δC 0.81 (3H, d, J = 7.2 Hz, H3-2′)/19.8 (C-2′), 0.78 (3H, d, J = 7.2 Hz, H3-3′)/18.2 (C-3′), 2.07 (1H, m, H-1′)/32.2 (C-1′), 4.18 (1H, d, J = 4.8 Hz, H-3)/60.1 (C-3) and δC 173.0 (C-2). In addition, signals for a 2-benzylmalonamide moiety were observed. The signals at δC 138.6 (C-2″), δH/δC 7.23 (2H, m)/130.2 (C-3″,7″), 7.24 (2H, m)/129.4 (C-4″,6″) and 7.14 (1H, m)/127.7 (C-5″) supported the existence of a phenyl group. In addition, the coupled signals at δH 3.11 (1H, dd, J = 13.8, 6.6 Hz, H-1″a) and 2.88 (1H, dd, J = 13.8, 8.4 Hz, H-1″b) showed vicinal coupling to the signal at δH 4.68 (1H, dd, J = 8.4, 6.6 Hz, H-6) in the 1H,1H-COSY spectrum. Furthermore, the two amidic carbonyls (C-5 and C-7) of the 2-benzylmalonamide moiety resonate at δC 173.2 and 173.1. Finally, a three-proton singlet at δH 1.90, which correlates with the 13C signal at δC 22.9, was assigned to a methyl group linked to N-1. The linkage of the valine residue with the 2-benzylmalonamide moiety through N-1 and N-4 was supported by the HMBC correlations of H-3/C-5, H-3/C-2, H3-8/C-2 (δC 173.0) and H3-8/C-7 (δC 173.1). The HMBC correlations of H3-8 to C-2 and C-7 secured the placement of the N-methyl at N-1. Additional HMBC correlations within the valine and the 2-benzylmalonamide moieties (Table 1 and Figure 2) confirmed the assignment of all carbon signals and completing the flat structure of 2. The absolute configuration of the chiral center at C-3 of the valine residue was established by analysis of the degradation product of 2. A small amount of 2 was hydrolyzed with 6N HCl to give valine residue, which was analyzed by chiral HPLC, and its retention time was compared with the retention times of authentic standards (d- and l-valine). The amino acid in 2 was determined as l-valine. In addition, the relative configuration of C-6 was determined as 6S*, since the NOESY spectrum (Table 1) exhibited a strong correlation between H-3 (δH 4.18) and H-6 (δH 4.68). The MM2 ChemBio3D Ultra 14.0 (ChemBioOffice® Ultra 14.0) energy minimized drawing for Compound 2 was also created and supports a significant NOESY correlation between H-3 and H-6 (Figure 3). Therefore, 2 was assigned as (3S,6S)-6-benzyl-3-isopropyl-1-methyl-1,4-diazepane-2,5,7-trione and named terretrione D. Terretrione D possesses the 1,4-diazepane skeleton, which was previously reported in terretriones A–C. These compounds were reported from the marine fungus Aspergillus terreus [18]. It is worth mentioning that terretrione D is 4-demethylterretrione B, and the NMR data of 2 are comparable with those of terretrione B [18]. Compound 2 is reported here for the first time from a natural source, and therefore, it is considered as a new natural product.
The known compounds were identified by extensive study of their spectral data, including HRESIMS, 1D and 2D NMR data, as well as by comparison with the available data in the literature. Thus, the compounds were identified as methyl-2-([2-(1H-indol-3-yl)ethyl]carbamoyl)acetate (1) [16], tryptamine (3), indole-3-carbaldehyde (4), 3,6-diisobutylpyrazin-2(1H)-one (5) [17] and terretrione C (6) [18]. The amino acid residue of compound 6 was also determined as N-methyl-l-isoleucine by HPLC chiral analysis of the hydrolysate of 6 (see Section 3.6). Furthermore, the relative configuration of its C-6 was assigned as 6S* by the NOESY correlation, similar to that described for 2.
Terretrione D (2): White amorphous powder; [α]D −65.7 (c 1.5, MeOH); UV (MeOH) λmax (log ε) 314 (2.65) nm; IR νmax (film) 3367, 1682, 1630, 1540, 1455, 1288, 1094, 702 cm−1; NMR data, see Table 1; HRESIMS m/z 289.1555 (calcd. for C16H21N2O3, [M + H]+, 289.1552).
see
http://www.mdpi.com/1660-3397/13/4/1698
Mar. Drugs 2015, 13(4), 1698-1709; doi:10.3390/md13041698
Article
Identification and Bioactivity of Compounds from the Fungus Penicillium sp. CYE-87 Isolated from a Marine Tunicate
1
Natural Products Unit, King Fahd Medical Research Center, King Abdulaziz University, Jeddah 21589, Saudi Arabia
2
Suez Canal University Hospital, Suez Canal University, Ismailia 51522, Egypt
3
Department of Natural Products, Faculty of Pharmacy, King Abdulaziz University, Jeddah 21589, Saudi Arabia
////////////  .
. .
.









































