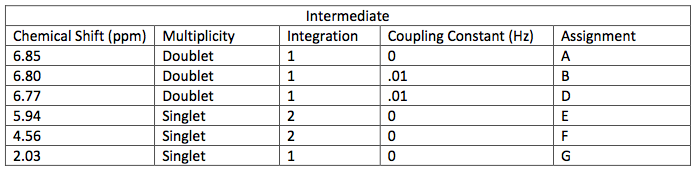
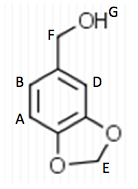
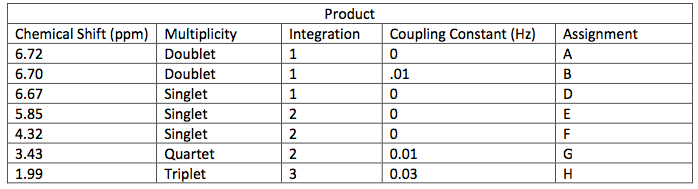
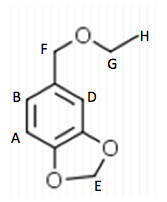
Starting material




Benzoin: Thaimine hydrochloride (1.52 g, 0.45 mmol), water (2mL) and 95% ethanol (15 mL) were combined in a 50-mL Erlenmeyer flask and swirled until dissolved and homogeneous. Aqueous sodium hydroxide (4.5 mL) was added and swirled until the solution appeared pale yellow. Finally, pure benzaldehyde (4.5 mL, 4.41 mmol) was added to the flask and the mixture was stored for two days. The crystals that formed at room temperature were placed in an ice bath and then filtered under vacuum. The crystals were washed with 5-mL portions of ice-cold water and left to dry. To isolate the pure product, the crude material was crystallized with 95% ethanol (24 mL). The pure product of benzoin showed the following physical characteristics: 2.07 g (44.3 % yield) mp: 129-132°C (lit: 135-135 °C). 1H NMR (CDCl3, 300 MHz) δ: 7.79 (d, J=1.5, 2H) 7.25 (m, 2H), 7.24 (m, 2H), 7.19 (m, 2H), 7.14 (m, 2H), 5.82 (d, J= 1.2, 1H), 3.92 (s, 1H) ppm. 13C NMR (CDCl3, 75Hz) δ: 199.2, 139.2, 139.1, 134.0, 129.2, 129.1, 128.7, 128.5, 127.8, 76.2 ppm. IR 3403, 3003, 1761, 1203 cm-1.
The final product of benzoin contained 13C NMR peaks at 199.2 ppm accounting for the carbonyl group and eight peaks in the range of 139.0-127.8 ppm representing the alkene bonds as well as the carbons of the aromatic rings. Finally, a peak at 76.2 ppm represented the carbon with the alcohol group attached. Regarding the1H NMR spectra, four multiplet peaks appeared in the range of 7.79 and 7.14 ppm representing the hydrogens surrounding the aromatic rings. A peak at 5.82 ppm accounted for the hydrogen attached to the carbon containing the alcohol group. A peak at 3.92 ppm represented the hydrogen of the alcohol group. An impurity of ethanol appeared at 4.42 ppm. Finally, the IR spectra displayed a peak at 3403 cm-1 representing the C-H stretches, a peak at 3003 cm-1, accounting for the alcohol group, and a strong peak at 1761 cm-1representing the carbonyl group. Overall, the spectra confirmed the condensation of benzoin.

Benzil: Benzoin (2.51g, 1.18 mmol), concentrated nitric acid (12 mL, 28.8 mmol), and a stir bar were placed into a 25-mL round-bottom flask with a water condenser and heated in a hot water bath at 70 °C for one hour. After heating and magnetically stirring, the mixture was added to 40 mL of cool water and stirred until crystallized into a yellow solid. Vacuum filtration was used to collect the crude product. The pure product was collected through recrystallization by using 95% ethanol (20 mL). The product displayed the following properties: 1.91 g (76.8 % yield) mp: 89-92 °C (lit: 95 °C). 1H NMR (CDCl3, 300 MHz) δ: 7.86 (m, 4H), 7.56 (m, 2H), 7.53 (m, 4H) ppm. 13C NMR (CDCl3, 75Hz) δ: 192.0, 132.3, 130.4, 127.3, 126.5 ppm. IR 3010 (w), 1668 (s) cm-1.
The 13C NMR produced a peak at 192.0 ppm representing the two carbonyl groups. Four peaks appeared between 132.3 and 126.5 ppm accounting for the carbons within the aromatic ring and the alkene bonds. The 1H NMR displayed three multiplet peaks at 7.86, 7.56, and 7.53 ppm representing the hydrogens around the aromatic ring that coupled with the surrounding hydrogens. Finally, the IR spectrum produced a C-H stretch peak at 3010 cm-1 and a carbonyl peak at 1668 cm-1. This data proved the success of the oxidation of benzoin to produce benzil.

1H-NMR: Benzil300 MHz, CDCl3delta [ppm]mult.atomsassignment7.50m (d)4 H2-H, 2'-H, 6-H, 6'-H7.64m (dd)2 H4-H, 4'-H7.95m (dd)4 H3-H, 3'-H, 5-H, 5'-H

| 13C-NMR: Benzil | |||
| 75.5 MHz, CDCl3 | |||
| delta [ppm] | assignment | ||
| 129.0 | CH arom. | ||
| 129.8 | CH arom. | ||
| 133.0 | CH arom. | ||
| 154.8 | C quart. arom. | ||
| 194.5 | C=O | ||
| 76.5-77.5 | CDCl3 | ||

IR: Benzil[KBr, T%, cm-1][cm-1]assignment3064arom. C-H valence1660C=O valence, ketone1594, 1579arom. C=C valence

GC: pure productcolumnDB-1, L=28 m, d=0.32 mm, film=0.25 µminleton column injection, 0.2 µLcarrier gasH2, 40 cm/soven90°C (5 min), 10°C/min --> 240°C (30 min)detectorFID, 270°Cintegrationpercent concentration calculated from relative peak area
Benzilic Acid: Benzil (2.10 g, 1.0 mmol), 95% ethanol (6 mL), and a boiling stone were added to a 25-mL round-bottom flask with a reflux condenser and heated until the solid benzil was dissolved. Aqueous potassium hydroxide (5 mL, 18.2 mmol) was added dropwise to the flask and the mixture was boiled for 15 minutes. The mixture was cooled, transferred to a beaker, and placed in an ice-water bath until crystallized. The crystals were isolated through vacuum filtration and washed with 4-mL portions of cold 95% ethanol. The solid was transferred to a 100-mL flask of hot water (60 mL) and mixed until completely dissolved. Concentrated hydrochloric acid (1.3 mL) was added drop-wise until a permanent solid was present and a pH of 2 was maintained. The solution was cooled in an ice bath and the crystals were filtered through vacuum filtration and washed with 2, 30-mL portions of ice-cold water. The remaining crystals were identified by the following properties: 0.41 g (17.4% yield) mp: 151-152 °C (lit: 150 °C). 1H NMR (CDCl3, 300 MHz) δ: 7.47 (m, 6H), 7.26 (s, 4H), 2.18 (s, 1H) ppm. 13C NMR (CDCl3, 75Hz) δ: 175.8, 141.4, 128.3, 128.2, 127.4, 82.0 ppm. IR 3399 (s), 2889 (s, b), 1718 (s), 1177 (s) cm-1.
The melting point corresponded to the known melting point of 150 °C. The 13C NMR spectra displayed a weak peak at 175.8 ppm, which accounted for the carbonyl group within the carboxylic acid. Four peaks at 141.4, 128.3, 128.2, and 127.4 ppm represented the carbons within the aromatic rings. Finally, a peak at 82.0 ppm represented the carbon attached to the alcohol group. The 1H NMR spectrum produced a peak at 7.47 and 7.26 ppm representing the two groups of equivalent hydrogens attached to the aromatic rings. A peak at 2.18 ppm represented the hydrogen of the alcohol group. A peak did not appear at 12 ppm that would have represented the hydrogen of the carboxylic group, which means the reaction was not carried to completion. In the IR spectrum, a hydroxyl peak appeared at 3399 cm-1. A broad peak appeared at 2889 cm-1 representing the carboxylic acid functional group of compound. Finally, a peak at 1718 cm-1 represented the carbonyl group and a peak at 1177 cm-1 accounted for the carbon-oxygen bond in both alcohol groups.



raman

1 Bruggink, A.; Schoevaart, R.; Kieboom, T. Org. Proc. Res. Dev., 2002, 7,622-640.
2 Pavia, L; Lampman, G; Kriz, G; Engel, R. A Small Scale Approach to Organic Laboratory Techniques, 2011, 266-269.
3 Lachman, A. J. Am. Chem. Soc., 1922, 44, 330-340.
extras




 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE

lahore, pakistan
 .
.
 .
.
 .
.








Punjabi sweets on a leaf in Lahore Pakistan, 20 Rupees or about 30 cents. I wasn't sure if one is supposed to eat the leaf too but I ate it anyways. 10/07



New Food Street Lahore
..............
extras
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus
 amcrasto@gmail.com
amcrasto@gmail.com
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus
amcrasto@gmail.com
lahore, pakistan
..Punjabi sweets on a leaf in Lahore Pakistan, 20 Rupees or about 30 cents. I wasn't sure if one is supposed to eat the leaf too but I ate it anyways. 10/07
New Food Street Lahore
..............


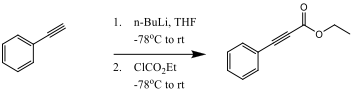
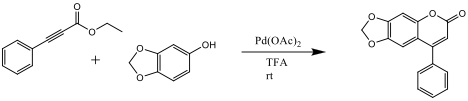
 The insertion of the ethyl phenylpropiolate to the sesamol-palladium intermediate is initially achieved in a cis confirmation. There is then an internal rearrangement of the palladium and CO2Et ligands to the trans confirmation which then allows for an electrophilic aromatic substitution to close the ring.
The insertion of the ethyl phenylpropiolate to the sesamol-palladium intermediate is initially achieved in a cis confirmation. There is then an internal rearrangement of the palladium and CO2Et ligands to the trans confirmation which then allows for an electrophilic aromatic substitution to close the ring.






 4-Bromo-1,2-(methylenedioxy)benzene
1H NMR
4-Bromo-1,2-(methylenedioxy)benzene
1H NMR 13 C NMR
13 C NMR
 IR
IR
 MASS
MASS
 RAMAN
RAMAN









 MASS
MASS
 IR
IR
 AND
AND




 NMR Spectroscopic Data-
NMR Spectroscopic Data-