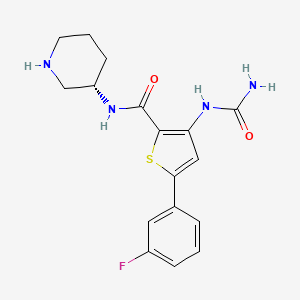
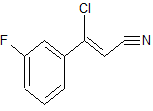
A multikilogram synthesis of AZD7762 has been achieved using a highly convergent route employing two efficient telescoped sequences to generate the key intermediates. Aminothiophene 11 is formed in a four-step, one-pot addition–elimination–cyclization sequence from cinnamonitrile 9, constructing the trisubstituted thiophene ring with the desired API substitution pattern in place. Cinnamonitrile 9 is derived by elaboration of 3-fluoroacetophenone. Generation of the urea function, followed by deprotection, affords AZD7762 in 49% yield over 5 isolated stages from chiral piperidine 5, a 5-fold increase in yield versus the first generation route, reducing the starting material burden and eliminating the previous requirement for metal-mediated couplings and chromatography.
Patent
formula I
I or a pharmaceutically acceptable salt thereof, comprising the following steps:
Example 1:
Synthesis of (Z)-3-Chloro-3-(3-fluorophenyl)-acrylonitrile from 3'-Fluoroacetophenone.
To a solution of 3'-fluoroacetophenone (80.0 g, 0.579 mol) in 7V,N-dimethyl formamide (560 ml) at about 400C was added phosphoryl chloride (92.50 ml, 1.01 mol) dropwise, maintaining the temperature at about 39-410C during the addition. The resulting reaction mixture was stirred at about 400C overnight before sampling for conversion to 2 by HPLC.
To the resulting reaction mixture was added a solution of hydroxylamine hydrochloride (45.17 g, 0.637 mol) in 7V,N-dimethyl formamide (240 ml) dropwise, maintaining the temperature at about 39-45°C during the addition, followed by a line -wash of JV,Λ/-dimethyl formamide (40 ml). After stirring at about 400C for 15 min, the reaction mixture was sampled for conversion to 4 before cooling to about 15-200C and addition of water (800 ml) dropwise, maintaining the temperature between about 17 to about 210C. The reaction mixture was then cooled to about 5°C and held at this temperature for a further 20 min before filtration of the solid, displacement washing with two separate portions of water (2 x 240 ml) and drying at about 400C overnight to afford the title compound as a pale yellow solid (74.24 g, 71% yield).
IH NMR (400MHz, DMSO-d6) δ: 7.72-7.65 (m, 2H), 7.63-7.56 (m, IH), 7.49-7.42 (m, IH),
7.03 (s, IH).
13C NMR (400MHz, DMSO-d6) δ: 162.0 (d, J = 245 Hz), 149.3 (d, J = 3 Hz), 135.6 (d, J= 8
Hz), 131.1 (d, J = 9 Hz), 123.3 (d, J = 3 Hz), 118.8 (d, J = 21 Hz), 115.8, 113.8 (d, J = 24 Hz),
89.3.
Example 2:
Synthesis of tert-buty\ (3S)-3-({[3-amino-5-(3-fluorophenyl)thiophen-2-yl]carbonyl}amino)piperidine-l-carboxylate from (S)-l-Boc-3-aminopiperidine and compound 4.

l-Boc-3-(S)-aminopiperidine (120.0 g, 0.599 mol) was dissolved in 2-methyltetrahydrofuran (540 ml). Pyridine (58.14 ml, 0.719 mol) was added, followed by a line-wash of 2-methyltetrahydrofuran (60 ml). Chloroacetyl chloride (55.32 ml, 0.689 mol) was added dropwise, maintaining the temperature at about 21-25°C, followed by a line wash of 2-methyltetrahydrofuran (60 ml). After 2.5 h at ambient temperature, the reaction mixture was sampled for conversion to 6 by HPLC before the addition of a 16% w/w aqueous solution of sodium chloride (360 ml). The mixture was stirred for 30 min before separating off the aqueous phase.
To the organic phase was added a filtered solution of potassium thioacetate (102.65 g, 0.899 mol) in water (204 ml), followed by a line-wash of water (36 ml), maintaining the temperature at about 19-26°C throughout. After stirring overnight at ambient temperature, the organic phase was sampled for conversion to 7 by HPLC before separating off the aqueous phase.
To the organic phase was added 4 (97.93 g, 0.539 mol) before dropwise addition of a solution of sodium methoxide in methanol (202 ml @ 25% w/w, 0.899 mol), maintaining the temperature at about 21-24°C. This was followed by a line wash of methanol (36 ml). After stirring for 1 h 50 min at ambient temperature, the reaction mixture was sampled by HPLC for conversion to 9 before heating to about 33°C, followed by dropwise addition of water (600 ml). After stirring for 10 min, the aqueous phase was separated off.
To the organic phase was added isohexane (960 ml) dropwise before removing a small sample of the reaction mixture, allowing it to cool and returning it to the bulk mixture to seed crystallisation. Dropwise addition of a second portion of isohexane (480 ml), followed by a ramped cool to about 3°C over 1 h and a subsequent hold at this temperature overnight caused crystallisation of the product. Filtration, displacement washing the solid with ice-cold tert-butyi acetate (240 ml) and 2 x ice-cold mixed solvent system of tert-butyl acetate and isohexane (1 :1, 2 x 240 ml) and drying at about 400C over 3 days afforded 9 as a pale yellow solid (192.69 g, 77% yield based on l-Boc-3-(S)-aminopiperidine).
IH NMR (400MHz, DMSO-d6, 8O0C) δ: 7.49-7.32 (m, 3H), 7.19-7.12 (m, IH), 7.01 (s, IH), 6.91 (d, IH), 6.29 (br, s, IH), 3.91-3.64 (m, 3H), 2.96-2.77 (m, 2H), 1.92-1.77 (m, IH), 1.74-1.30 (m, 12H). Mass Spectrum: 420 [MH]+ and 364 [M-Φu]+.
Example 3:
Synthesis of tert-butγ\ (3S)-3-({[5-(3-fluorophenyl)-3- { [(trichloroacetyljcarbamoyl] amino}thiophen-2-yl] carbonyl}amino)piperidine-l-carboxylate from compound 9 and trichloroacetyl isocyanate.
To a solution of 9 (73.12 g, 0.174 mol) in tetrahydrofuran (800 ml) was added trichloroacetyl isocyanate (23.23 ml, 0.196 mol), maintaining the temperature at about 20-300C during the addition. After 2.5 h at ambient temperature, the mixture was sampled for conversion to 10 before addition of isohexane (1120 ml) dropwise over 1 hour. After stirring for a further 1 h, the reaction mixture was filtered, the solid washed with isohexane (160 ml) and dried at about 400C to afford 10 as a pale peach solid (103.54 g, 98% yield).
IH NMR (400MHz, DMSO-d6, 702C) δ: 11.70 (s, IH), 11.49 (br. s, IH), 8.24 (s, IH), 7.80 (d, IH), 7.57-7.40 (m, 3H), 7.26-7.18 (m, IH), 3.97-3.67 (m, 3H), 2.95-2.78 (m, 2H), 1.97-1.84 (m, IH), 1.78-1.53 (m, 2H), 1.51-1.33 (m, 10H).
13C NMR (400MHz, DMSO-d6) δ: 162.3 (d, J = 245 Hz), 161.7, 160.3, 153.7, 148.5, 141.9 (d, J = 3 Hz), 140.5, 134.6 (d, J = 8 Hz), 131.1 (d, J = 9), 121.4 (d, J = 3 Hz), 119.5, 115.3 (d, J = 21 Hz), 114.7, 112.0 (d, J = 23 Hz), 91.8, 78.4, 47.4, 45.7, 43.2, 29.2, 27.7, 23.2.
Example 4:
Synthesis of tert-butγ\ (3S)-3-({[3-(ureido)-5-(3-fluorophenyl)thiophen-2-yl]carbonyl}amino)piperidine-l-carboxylate via deprotection of compound 10.
To a suspension of 10 (101.45 g, 0.169 mol) in methanol (516 ml) was added triethylamine (58.15 ml, 0.417 mol). After a further 2.5 h at ambient temperature, the mixture was sampled for conversion to 11 before addition of water (206 ml) over 10 min. After stirring overnight at ambient temperature, the reaction mixture was heated to about 45°C for 15 min before addition of a second portion of water (1083 ml) over 2 h. After a further 1 h at about 45°C, the reaction mixture was allowed to cool to about 200C and held at this temperature for 1 h. The reaction mixture was filtered and the solid washed with water (206 ml) before drying at about 400C overnight to afford 10 as a white solid (77.10 g, 99% yield).
IH NMR (400MHz, DMSO-d6, 8O0C) δ: 9.86 (s, IH), 8.24 (s, IH), 7.60-7.41 (m, 3H), 7.41-7.33 (m, IH), 7.22-7.15 (m, IH), 6.36 (br, s, 2H), 3.94-3.68 (m, 3H), 2.97-2.79 (m, 2H), 1.94-1.84 (m, IH), 1.76-1.55 (m, 2H), 1.47-1.34 (m, 10H) Mass Spectrum: 486 [MNa]+.
Example 5:
Synthesis of 5-(3-Fluorophenyl)-3-ureidothiophene-2-carboxylic acid (S)-piperidin-3-ylamide via deprotection of compound 11.
11 12
To a suspension of 11 (75.3 g, 0.163 mol) in methanol (383 ml) was added an aqueous solution of hydrochloric acid (40.78 ml @ 37% w/w in water, 0.488 mol) dropwise, maintaining the temperature at about 20-300C. The resulting reaction mixture was then heated at about 500C for 4 h before sampling for conversion to 12. Triethylamine (85.10 ml, 0.610 mol) was added dropwise before addition of water (345 ml). A small sample of the reaction mixture was then removed, allowing it to cool before returning to the bulk mixture to seed crystallisation with stirring for 30 min. Further water (613 ml) was added over 1.5 h before holding at about 500C for a further 30 min and allowing to cool to about 200C with stirring overnight. The reaction mixture was filtered and the solid washed with water (153 ml) before drying at about 400C overnight to afford 12 as a white solid (57.26 g, 97% yield).
IH NMR (400MHz, DMSO-d6, 8O0C) δ: 9.88 (br. s, IH), 8.22 (s, IH), 7.52-7.36 (m, 4H), 7.19 (m, IH), 6.35 (br. s, 2H), 3.81 (m, IH), 2.95 (m, IH), 2.76 (m, IH), 2.44-2.56 (m, 2H), 1.82 (m, IH), 1.67-1.34 (m, 3H). Mass Spectrum: 363 [MH]+.
Example 6:
Purification of 5-(3-Fluorophenyl)-3-ureidothiophene-2-carboxylic acid (S)-piperidin-3-ylamide (compound 12).
A suspension of 12 (50.0 g, 0.138 mol) in methanol (650 ml) was heated to about 300C for 30 min before filtering the resulting hazy suspension through a 1.6 micron glass microfibre filter paper into a second vessel, followed by a line-wash with methanol (100 ml), discarding the solid residue. The resulting solution was cooled to about 100C before addition of water (250 ml), dropwise over 20 min, maintaining the temperature at about 10-150C. To seed crystallisation, a sample of purified 12 was then added (150 mg, 0.3% wt/wt), and the contents of the vessel allowed to stir at about 100C for 30 min. Addition of a second portion of water (500 ml) over 1 h 30 min, maintaining the temperature at about 10-130C, followed by stirring for 20 h at about 100C, resulted in complete crystallisation. Filtration, washing the solid with water (2 x 100 ml), sucking dry for 30 min before drying under vacuum at about 400C overnight, afforded purified 12 as a white solid (46.91 g, 92% yield).
IH NMR (400MHz, DMSO-d6) δ: 10.04 (s, IH), 8.29 (s, IH), 7.77 (d, IH), 7.55 - 7.42 (m, 3H), 7.24 (m, IH), 6.67 (br. s, 2H), 3.79 (m, IH), 2.94 (m, IH), 2.78 (m, IH), 2.49 - 2.37 (m, 2H), 1.82 (m, IH), 1.65 - 1.34 (m, 3H). Mass Spectrum: 363 [MH]+.
Example 7:
Synthesis of 5-(3-Fluorophenyl)-3-ureidothiophene-2-carboxylic acid (S)-piperidin-3-ylamide fumarate salt (compound 12 Fumarate salt).


12 Fumarate salt To a mixture of 12 (1.00 g, 2.8 mmol) and fumaric acid (160 mg, 1.4 mmol) was added acetone (3.0 ml) and water (1.9 ml). The resulting hazy solution was filtered through a syringe filter, before adding it dropwise to a second vessel containing a solution of fumaric acid (160 mg, 1.4 mmol) in acetone (18.5 ml) and water (0.5 ml), and a seed crystal of 12 Fumarate salt. The solution addition took place at ambient temperature over 1 h and was followed by a line -wash with acetone (1.0 ml) and water (0.1 ml). Gradual crystallisation of the product occurred, and after stirring the resulting slurry at ambient temperature for 1 h 30 min, the solid was filtered and washed with acetone (2 x 2.0 ml), sucking dry for 30 min before drying under vacuum at about 40
0C overnight to afford 12 Fumarate salt as a white solid (0.96 g, 96% yield).
IH NMR (400MHz, DMSO-d6) δ: 10.00 (s, IH), 8.29 (s, IH), 8.24 (d, IH), 7.54 - 7.42 (m, 3H), 7.24 (m, IH), 6.67 (br. s, 2H), 6.52 (s, 2H [2 H Fumaric acid]), 4.16 (br. m, IH), 3.22 (m, IH), 3.09 (m, IH), 2.91 - 2.76 (m, 2H), 1.86 (m, 2H), 1.65 (m, 2H). Mass Spectrum: 363 [MH]+.
Example 8:
Synthesis of 5-(3-Fluorophenyl)-3-ureidothiophene-2-carboxylic acid (S)-piperidin-3-ylamide fumarate salt (compound 12 Hemi-Fumarate salt).
12 12 Hemi-Fumarate salt
To a solution of 12 (2.0 g, 5.6 mmol) in methanol (33.7 ml) was added fumaric acid (327 mg, 2.8 mmol) and the resulting solution was stirred for 30 min at about 18°C. After seeding the solution with 12 Hemi-Fumarate salt (5 mg, 0.006 mmol) and stirring for 5 h at about 18-19°C, the reaction mixture was cooled to about 5°C, stirring was ceased and the reaction was held at this temperature overnight. Filtration of the resulting solid, washing with methanol (1 x 2 ml) and sucking dry on the filter afforded 12 Hemi-Fumarate salt as a white solid (1.90 g, 80%).
IH NMR (400MHz, DMSO-d6) δ: 10.02 (s, IH), 8.28 (s, IH), 8.08 (d, IH), 7.54 - 7.42 (m, 3H), 7.24 (m, IH), 6.66 (br s., 2H), 6.47 (s, IH [2 H Fumaric acid]), 4.02 (br. m, IH), 3.11 (m, IH), 2.96 (m,lH), 2.75 - 2.60 (m, 2H), 1.85 (m, IH), 1.76 (m, IH), 1.58 (m, 2H). Mass Spectrum: 363 [MH]+.