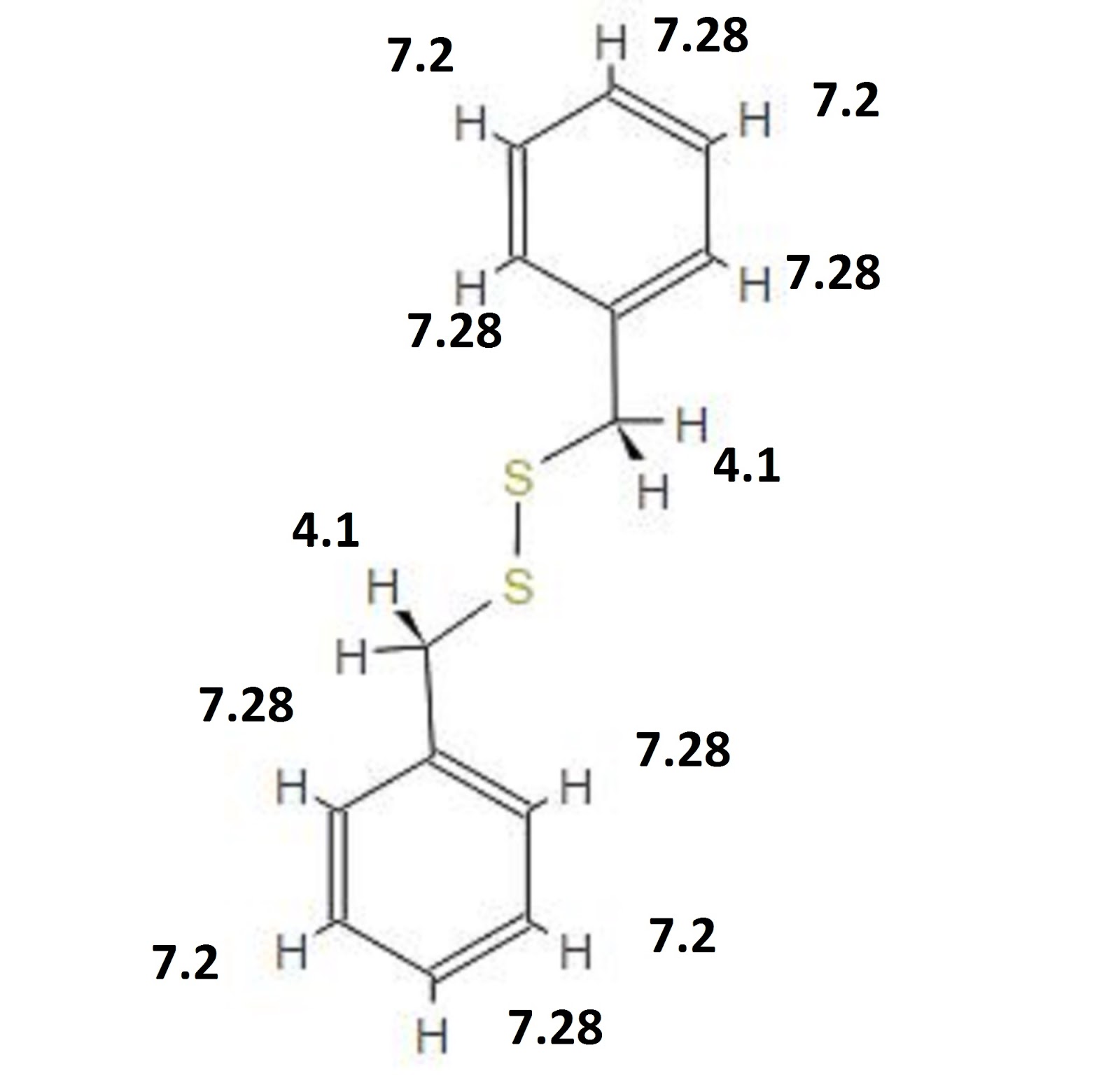

Figure 3. (a) Single ion mobility spectrum for ranitidine (m/z 315.2), (b) total ion mobility spectrum, (c) combined mass spectra from ranitidine IMS peak (scans 2029 - 2032), and (d) combined mass spectra from full IMS data (scans 2000 - 2200).

Ranitidine
Ranitidine, sold under the trade name Zantac among others, is a medication that decreases stomach acid production.[1] It is commonly used in treatment of peptic ulcer disease, gastroesophageal reflux disease, and Zollinger–Ellison syndrome.[1] There is also tentative evidence of benefit for hives.[2] It can be taken by mouth, by injection into a muscle, or into a vein.[1]
Common side effects include headaches and pain or burning if given by injection. Serious side effects may include liver problems, a slow heart rate, pneumonia, and the potential of masking stomach cancer.[1] It is also linked to an increased risk ofClostridium difficile colitis.[3] It is generally safe in pregnancy. Ranitidine is an H2 histamine receptor antagonist that works by blocking histamine and thus decreasing the amount of acid released by cells of the stomach.[1]
Ranitidine was discovered in 1976 at Glaxo Pharmaceuticals, now a part of GlaxoSmithKline.[4][5] It is on the World Health Organization's List of Essential Medicines, the most important medications needed in a basic health system.[6] It is available as a generic medication.[1] The wholesale price in the developing world is about 0.01 to 0.05 USD per pill.[7] In the United States it is about 0.05 USD per dose.[1]



Laboratory Synthesis Of Ranitidine
Synthesis Of Ranitidine
---------------------------------------------------------------------------------------

Ranitidine Synthetic procedure/method of synthesis
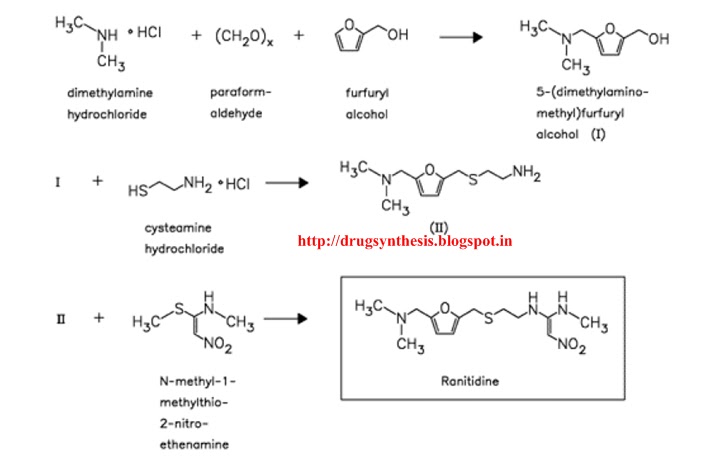
The reaction of 5-dimethylaminomethyl-2-furanylmethanol (I) with 2-mercaptoethylamine (II) by means of aqueous HCl gives 2-[[(5-dimethylamino-methyl-2-furanyl)methylthio]ethaneamine (III), which is then condensed with N-methyl-1-methylthio-2-nitrotheneamine (IV) by heating at 120 C. Compound (IV) is obtained by reaction of 1,1-bis(methylthio)-2-nitroethene (V) with methylamine in refluxing ethanol
Ranitidine reference
- Serradell, M.N.; Blancafort, P.; Casta馿r, J.; Hillier, K.; Ranitidine. Drugs Fut 1979, 4, 9, 663
- Price, B.J. et al. (Allen and Hanburys, Ltd.); US 4128658.
- Price, B.J.; Bradshaw, J.; Clitherow, J.W. (Allen & Hansburys Ltd.); Aminoalkyl furan derivatives.. DE 2734070; FR 2360587; US 4128658 ,DE 2734070; FR 2360587; US 4128658.
PAPER
Synthesis of ranitidine (Zantac) from cellulose-derived 5-(chloromethyl)furfural
*Corresponding authors
aDepartment of Chemistry, University of California Davis, 1 Shields Avenue, Davis, US
E-mail: mascal@chem.ucdavis.edu
Fax: 530-752-8995
Tel: 530-754-5373
E-mail: mascal@chem.ucdavis.edu
Fax: 530-752-8995
Tel: 530-754-5373
Green Chem., 2011,13, 3101-3102
DOI: 10.1039/C1GC15537G
The biomass-derived platform chemical 5-(chloromethyl)furfural is converted into the blockbuster antiulcer drug ranitidine (Zantac) in four steps with an overall 68% isolated yield.



PROCESS

2. Experimental Procedures
5-[[(2-Acetamidoethyl)thio]methyl]furfural 14
Sodium hydride (95%) (103 mg, 4.08 mmol) was added to a solution of Nacetylcysteamine (0.4051 g, 3.40 mmol) in dry THF (20 mL) under argon. The resulting suspension was stirred at RT for 30 min and a solution of CMF 12 (0.4912 g, 3.40 mmol) in dry THF (10 mL) was added dropwise over a 10 min period. The resulting light yellow solution was allowed to stir overnight at RT. The solvent was evaporated and saturated brine (50 mL) was added. The mixture was extracted with CH2Cl2 (2 × 50 mL) and the organic layers were combined and washed with saturated brine (100 mL). The organic layer was dried over Na2SO4. Charcoal (100 mg) was added and the mixture was stirred for 20 min and filtered. The solvent was evaporated to give 14 as a yellow liquid (0.7042 g, 91 %). 1H NMR (CDCl3, 300 MHz) 9.58 (1H, s), 7.21 (1H, d, J = 3.6 Hz), 6.48 (1H, s, br), 5.95 (1H, d, J = 3.6 Hz), 3.79 (2H, s), 3.45 (2H, q, J = 6.3 Hz), 2.72 (2H, t, J = 6.6 Hz), 2.00 (3H, s); 13C NMR (CDCl3, 75 MHz) 23.1, 27.8, 31.7, 38.4, 110.7, 121.9, 152.2, 158.9, 170.7, 177.4; IR (neat) 3298, 3101, 1663, 1548, 1512, 1287, 1022, 772 cm-1; HRMS (ESI): calculated for C10H14O3NS: [M+H]+ 228.0694: found 228.0690.
5-[[(2-Acetamidoethyl)thio]methyl]-N,N-dimethyl-2-furanmethanamine 15
Me2NH (1.0 mL) was added to a solution of 14 (0.2105 g, 0.926 mmol) in dry methanol (20 mL) and the mixture was stirred at RT for 1 h. The resulting red solution was cooled to 0 °C and NaBH4 (98 %) (55 mg, 1.42 mmol) was added over a 5 min period. The mixture was allowed to come to RT and stirred for 30 min. The solvent was evaporated while keeping the bath temperature below 45 °C. The residue was dissolved in CH2Cl(50 mL) and filtered to remove inorganic impurities. The solvent was evaporated to give 15 (0.2145 g, 90 %) as a pale yellow oil. 1H NMR (CDCl3, 300 MHz) 6.42 (1H, s, br), 6.09 (1H, s), 3.67 (2H, s), 3.37 (2H, s), 3.26 (2H, q, J = 6.0 Hz), 2.62 (2H, t, J = 6.4 Hz) 2.21 (6H, s), 1.93 (3H, s); 13C NMR (CDCl3, 75 MHz) 23.5, 28.4, 31.9, 38.7, 45.4, 56.2, 108.4, 109.9, 151.4, 152.1, 170.5; IR (neat) 3273, 2944, 1656, 1545, 1291, 1019, 729 cm- 1 ; HRMS (ESI): calculated for C12H21O2N2S: [M+H]+ 257.1322: found 257.1323.
5-[[(2-aminoethyl)thio]methyl]-N,N-dimethyl-2-furanmethanamine 5
A solution of 15 (0.2473 g, 0.965 mmol) in freshly prepared 2N aq NaOH (10 mL) was heated at reflux for 2 h. The mixture was cooled to RT and extracted with CH2Cl2 (3×30 mL). The organic layers were combined and washed with saturated brine, dried over Na2SO4, and evaporated to give 5 (0.1934 g, 94 %) as a pale yellow oil. 1H NMR (CDCl3, 300 MHz) 6.02 (2H, s), 3.61 (2H, s), 3.33 (2H, s), 2.74 (2H, t, J = 6.3 Hz), 2.52 (2H, t, J = 6.6 Hz), 2.16 (6H, s); 13C NMR (CDCl3, 75 MHz) 28.2, 35.9, 40.9, 45.1, 55.9, 108.1, 109.5, 151.4, 152.1; IR (neat) 3359 cm-1, 2947, 2769, 1559, 1459, 1015, 797 cm-1; HRMS (ESI): calculated for C10H19ON2S: [M+H]+ 215.1212: found 215.1218.
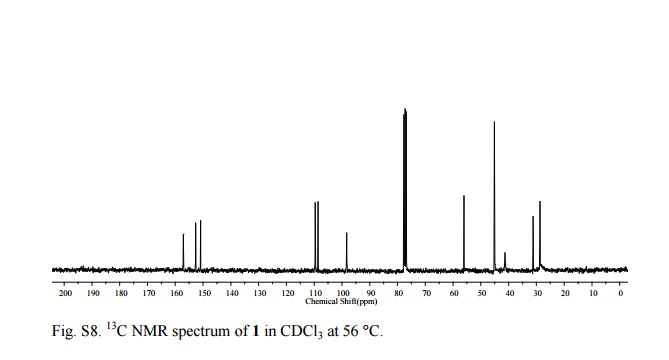
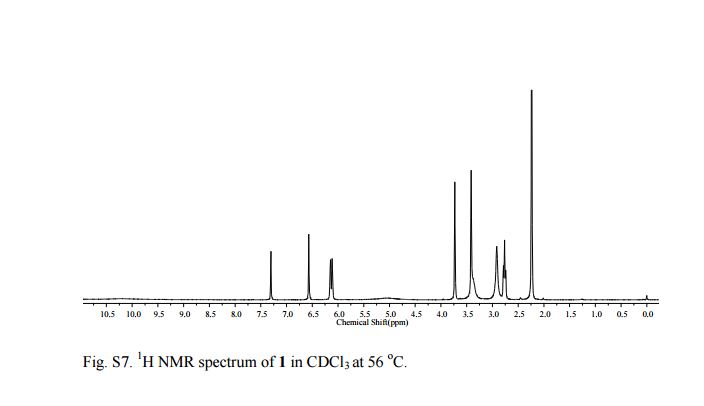
N-[2-[[[5-[(dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-N'-methyl-2-nitro- 1-Ethenediamine (Ranitidine) 1 The experimental procedure is modified from existing literature:2 A solution of 5 (0.1501 g, 0.700 mmol ) in distilled water (10 mL) was added dropwise over a period of 10 min to a suspension of 1-methylthio-1-methylamino-2-nitroethylene 7 (0.1041 g, 0.703 mmol) in distilled water (5 mL) with stirring. The resulting light yellow solution was placed in an oil bath at 55 °C and the mixture was stirred at that temperature overnight. Saturated brine (30 mL) was added and the mixture was extracted with CHCl3 (3×20 mL). The combined organic layer was dried over Na2SO4. Evaporation of the solvent gave 1 as a pale yellow oil (0.1935 g, 88 %). 1H NMR (CDCl3, 300 MHz, 56 oC) 10.23-10.15 (1H, br, NH), 6.57 (1H, s), 6.13 (2H, d, 6.0 Hz), 5.04 (1H, br, NH), 3.73 (2H, s), 3.41 (4H, s), 2.92 (2H, s), 2.76 (2H, t, 6.0 Hz), 2.24 (6H, s); 13C NMR (CDCl3, 75 MHz, 56 °C) 28.2, 30.6, 40.7, 44.6, 55.6, 97.9, 108.1, 109.1, 150.4, 152.1, 156.6; IR (neat) 3209, 2944, 2815, 2776, 1620, 1574, 1384, 1230, 1019, 761 cm-1; HRMS (ESI): calculated for C13H23O3N4S: [M+H]+ 315.1491: found 315.1497.

Zantac (ranitidine) 300-mg tablet
PATENT

Patent EP0796256B1 - Process for preparing ranitidine - Google Patents
Figure 00060001
HPLC

An Improved HPLC Method for the Determination of Ranitidine ...
An Improved HPLC Method for the Determination of Ranitidine Suitable for All Dosage Forms
PATENT

Figure imgf000004_0001
CLIP

CLIP
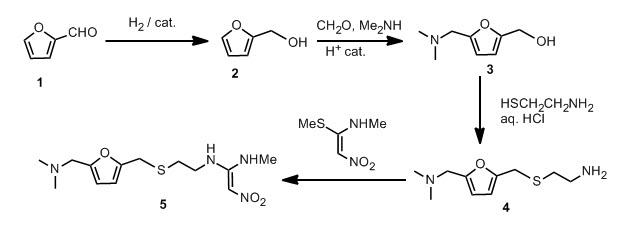
The paper was found in Green Chemistry,“Synthesis of ranitidine (Zantac) from cellulose-derived 5-(chloromethyl)furfural” by Mark Mescal et al, Green Chemistry, 2011,13, 3101-3102, DOI: 10.1039/c1gc15537g. Once again, I am beating the press before they print so I supplied the Digital Object Identifier. I am sure the sales for Ranitidine are quite large; who doesn’t get heartburn at one time or another. I think it is very fortunate the author shows you can use a starting material that can be derived from just about any source of cellulose. I find it interesting how renewable feedstocks can be utilized in industry and become part of important commodities, such as plastics, pharmaceuticals, etc. This paper refers to another discussing where the starting material was derived from. Starting material can be sugars, cellulose or raw cellulosic biomass and the reaction can produce yields of 80-90 %. M.Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 47, 7924; On with the show, though. The original synthetic route was provided in the paper and I will provide it to you.
On with the show, though. The original synthetic route was provided in the paper and I will provide it to you.
On with the show, though. The original synthetic route was provided in the paper and I will provide it to you.
Furfural 1 was reduced to give the furfuryl alcohol 2. The furfuryl alcohol is methylaminated to give 3, which is reacted with cysteamine in concentrated HCl to give 4. This is condensed with 1-methylthio-1-methylamino-2-nitroethylene to give the final product. The patent literature has the yield < 50 % for the aminomethylation and subsequent reaction with cysteamine, but recently, these steps have been reported to have higher conversions.

This new synthesis, apart from using a renewable feedstock as a starting material, has synthetic steps with an average yield of 91 %, and requires no chromatography. Note that N-acetylcysteamine was used as opposed to cysteamine in the first step, in high yield. A reductive amination with methylamine gives 8 again in high yield. Treatment with KOH provides the free amine 9 and the final step is the condensation with the nitroethylene used in the previous synthesis
Paper
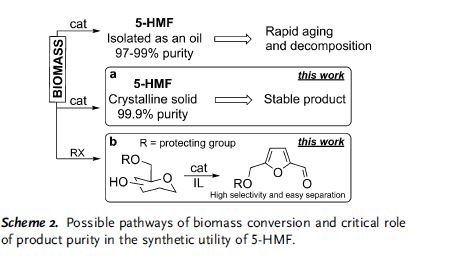
Critical influence of 5-hydroxymethylfurfural aging and decomposition on the utility of biomass conversion in organic synthesis
Angewandte Chemie, International Edition (2016), 55, (29), 8338-8342
Angewandte Chemie, International Edition (2016), 55, (29), 8338-8342

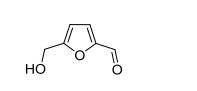
5-HMF. 1H NMR (400 MHz, DMSO-d6) δ = 9.54 (s, 1H, C(O)H), 7.49 (d, J = 3.5 Hz, 1H, CHfuran), 6.60 (d, J = 3.5 Hz, 1H, CH-furan), 5.57 (t, J = 5.9 Hz, 1H, OH), 4.51 (d, J = 5.9 Hz, 2H, CH2OH). 13C{1H} NMR (101 MHz, DMSO-d6) δ = 177.9 (C(O)H), 162.2, 151.7 (C-furan), 124.4, 109.7 (CH-furan), 55.9 (CH2OH). Anal. calcd. For C6H6O3 (126.11): C 57.14, H 4.80; found: C 57.08, H 4.79.
Abstract
Spectral studies revealed the presence of a specific arrangement of 5-hydroxymethylfurfural (5-HMF) molecules in solution as a result of a hydrogen–bonding network, and this arrangement readily facilitates the aging of 5-HMF. Deterioration of the quality of this platform chemical limits its practical applications, especially in synthesis/pharma areas. The model drug Ranitidine (Zantac®) was synthesized with only 15 % yield starting from 5-HMF which was isolated and stored as an oil after a biomass conversion process. In contrast, a much higher yield of 65 % was obtained by using 5-HMF isolated in crystalline state from an optimized biomass conversion process. The molecular mechanisms responsible for 5-HMF decomposition in solution were established by NMR and ESI-MS studies. A highly selective synthesis of a 5-HMF derivative from glucose was achieved using a protecting group at O(6) position.
PAPER
Phytochemical screening and investigation of antiulcer activity of Tridax procumbens
International Journal of Pharmacy and Technology (2015), 6, (4), 7679-7690
International Journal of Pharmacy and Technology (2015), 6, (4), 7679-7690
Lavanya Asula* , A. Sony John, Deepthi Kotturi, P. Srividyalaxmi, R. Soni and Y. Mamatha Kalyani Department of Pharmacy, Jawaharlal Nehru Technological University, Holy Mary Institute of Technology and Science College of Pharmacy Hyderabad, India. Email: lavanya.asula@gmail.com
PATENT
Waste gas treatment and methyl mercaptan recovery process in production process of cimetidine and ranitidine
cimetidine and ranitidine terms widely used in the treatment of stomach is bound to promote the continuous mass production of APIs, however, the raw material in the manufacturing process of the drug inevitably produce methyl mercaptan, dimethyl sulfide, a methylamine, carbon disulfide and nitromethane workshop emissions. Because of methyl mercaptan, dimethyl sulfide into the atmosphere having foul odor. Resulting in the production shop around smelling, and even affect the normal life of residents of several kilometers around. So some manufacturers use incineration method expects to dispose of the waste gas combustion, which reduces air pollution to some extent. But using incineration method has two drawbacks: one gas methyl mercaptan, dimethyl sulfide gas combustion higher value produce a few meters of flames burning heat generated while it is easy to burn incinerator, security posed by the chemical production big risk; on the other hand by a combustion method can not solve the odor problem, air pollution is still grim, because incomplete combustion, odor difficult to eliminate people's sense of smell is particularly sensitive to the perception of mercaptans, while burning a large amount of sulfur dioxide in the same air pollution. There's manufacturers to adopt authoritarian incinerator burning after the first use of chlorine dioxide generator eliminate odor, although this method has a certain smell to eliminate the effect of improving, but requires authoritarian equipment, increasing the cost of gas treatment and discharge sulfur dioxide into the air is still there.
PATENT
CN 102408398

Title: Ranitidine
CAS Registry Number: 66357-35-5
CAS Name: N-[2-[[[-5-[(Dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-N¢-methyl-2-nitro-1,1-ethenediamine
Molecular Formula: C13H22N4O3S
Molecular Weight: 314.40
Percent Composition: C 49.66%, H 7.05%, N 17.82%, O 15.27%, S 10.20%
Literature References: Histamine H2-receptor antagonist which inhibits gastric acid secretion. Prepn: B. J. Price et al., FR2384765; eidem, US 4128658 (both 1978 to Allen & Hanburys). HPLC determn in plasma: P. F. Carey, L. E. Martin, J. Liq. Chromatogr. 1979, 1291. Pharmacological studies: J. Bradshaw et al., Br. J. Pharmacol. 66, 464 (1979); M. J. Daly et al., Gut 21,408 (1980). Efficacy in treatment of duodenal ulcers: A. Berstad et al., Scand. J. Gastroenterol. 15, 637 (1980); R. P. Walt et al.,Gut 22, 49 (1981). Review of pharmacology and therapeutic use: R. N. Brogden et al., Drugs 24, 267-303 (1982). Comprehensive description: M. Hohnjec et al., Anal. Profiles Drug Subs. 15, 533-561 (1986).
Properties: Solid, mp 69-70°.
Melting point: mp 69-70°
Derivative Type: Hydrochloride
CAS Registry Number: 66357-59-3
Manufacturers' Codes: AH-19065
Trademarks: Azantac (GSK); Melfax (Apotex); Noctone (GEA); Raniben (Firma); Ranidil (Menarini); Raniplex (Fournier); Sostril (Cascan); Taural (Roemmers); Terposen (Vir); Trigger (Polifarma); Ulcex (Guidotti); Ultidine (GSK); Zantac (GSK); Zantic (GSK)
Molecular Formula: C13H22N4O3S.HCl
Molecular Weight: 350.86
Percent Composition: C 44.50%, H 6.61%, N 15.97%, O 13.68%, S 9.14%, Cl 10.10%
Properties: Off-white solid, mp 133-134°. Freely sol in acetic acid and water, sol in methanol, sparingly sol in ethanol. Practically insol in chloroform.
Melting point: mp 133-134°
Derivative Type: Bismuth citrate
CAS Registry Number: 128345-62-0
Additional Names: Ranitidine bismutrex
Manufacturers' Codes: GR-122311X
Trademarks: Pylorid (GSK); Tritec (GSK)
Molecular Formula: C13H22N4O3S.C6H5BiO7
Molecular Weight: 712.48
Percent Composition: C 32.03%, H 3.82%, N 7.86%, O 22.46%, S 4.50%, Bi 29.33%
Literature References: Pharmacology and activity vs Helicobacter sp: R. Stables et al., Aliment. Pharmacol. Ther. 7, 237 (1993).
Therap-Cat: Antiulcerative.
Keywords: Antiulcerative; Histamine H2-Receptor Antagonist.
References
- ^ Jump up to:a b c d e f g "Ranitidine". The American Society of Health-System Pharmacists. Retrieved Dec 1, 2015.
- Jump up^ Fedorowicz, Z; van Zuuren, EJ; Hu, N (14 March 2012). "Histamine H2-receptor antagonists for urticaria.". The Cochrane database of systematic reviews. 3: CD008596.doi:10.1002/14651858.CD008596.pub2. PMID 22419335.
- Jump up^ Tleyjeh, IM; Abdulhak, AB; Riaz, M; Garbati, MA; Al-Tannir, M; Alasmari, FA; Alghamdi, M; Khan, AR; Erwin, PJ; Sutton, AJ; Baddour, LM (2013). "The association between histamine 2 receptor antagonist use and Clostridium difficile infection: a systematic review and meta-analysis.". PLOS ONE. 8 (3): e56498. doi:10.1371/journal.pone.0056498.PMC 3587620
 . PMID 23469173.
. PMID 23469173. - Jump up^ Fischer, Janos (2010). Analogue-based Drug Discovery II. John Wiley & Sons. p. 4.ISBN 9783527632121.
- Jump up^ Hara, Takuji (2003). Innovation in the pharmaceutical industry the process of drug discovery and development. Cheltenham, U.K.: Edward Elgar. p. 94.ISBN 9781843765660.
- Jump up^ "WHO Model List of EssentialMedicines" (PDF). World Health Organization. October 2013. Retrieved 22 April 2014.
 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| N-(2-[(5-[(dimethylamino)methyl]furan-2-yl)methylthio]ethyl)-N'-methyl-2-nitroethene-1,1-diamine | |
| Clinical data | |
| Pronunciation | /rəˈnɪtᵻdiːn/ |
| Trade names | Zantac, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601106 |
| License data |
|
| Pregnancy category | |
| Routes of administration | Oral, IV |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 39 to 88% |
| Protein binding | 15% |
| Metabolism | Hepatic: FMOs, including FMO3; other enzymes |
| Biological half-life | 2–3 hours |
| Excretion | 30–70% Renal |
| Identifiers | |
| CAS Number | 66357-35-5 |
| ATC code | A02BA02 (WHO) A02BA07 (WHO) (ranitidine bismuth citrate) |
| PubChem | CID 3001055 |
| IUPHAR/BPS | 1234 |
| DrugBank | DB00863 |
| ChemSpider | 4863 |
| UNII | 884KT10YB7 |
| KEGG | D00422 |
| ChEBI | CHEBI:8776 |
| ChEMBL | CHEMBL1790041 |
| Synonyms | Dimethyl [(5-{[(2-{[1-(methylamino)- 2-nitroethenyl]amino}ethyl)sulfanyl] methyl}furan-2-yl)methyl]amine |
| Chemical data | |
| Formula | C13H22N4O3S |
| Molar mass | 314.4 g/mol |
//////////
/////////

 July-August 2012, Volume 2, No.4
July-August 2012, Volume 2, No.4