1,3-Diphenylisoquinoline (3a). Pale-yellow solid (103.5 mg,
92%),
mp 78-79 oC (lit.24, 73-74.5 oC). 24 J. D. Tovar and T. M. Swager, J. Org. Chem., 1999, 64, 6499
1H NMR (500 MHz, CDCl3)
δ 8.25-8.23 (m, 2H), 8.15-8.14 (m, 1H), 8.09 (s, 1H), 7.95-7.93
(m, 1H), 7.84-7.83 (m, 2H), 7.70-7.67 (m, 1H), 7.59-7.50 (m, 6H),
7.44-7.40 (m, 1H);
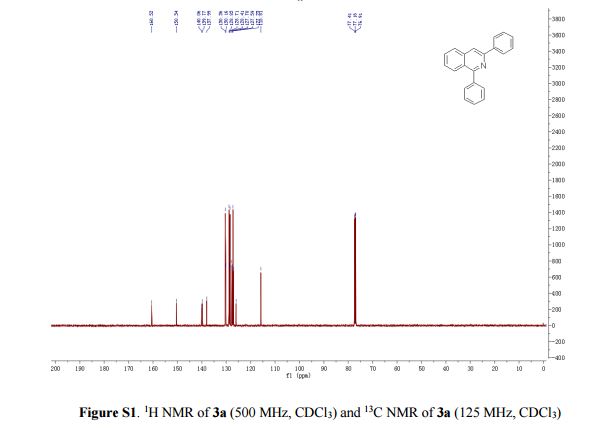
13C NMR (125 MHz, CDCl3) δ 160.5, 150.3,
140.1, 139.8, 138.0, 130.4, 130.2, 128.8, 128.7, 128.6, 128.4,
127.7, 127.6, 127.2, 127.0, 126.0, 115.8.

Efficient synthesis of isoquinolines in water by a Pd-catalyzed tandem reaction of functionalized alkylnitriles with arylboronic acids
Green Chem., 2017, Advance Article
DOI: 10.1039/C7GC00267J, Paper
Kun Hu, Linjun Qi, Shuling Yu, Tianxing Cheng, Xiaodong Wang, Zhaojun Li, Yuanzhi Xia, Jiuxi Chen, Huayue Wu
Pd-catalyzed tandem reaction of functionalized alkylnitriles with arylboronic acids for the synthesis of diverse isoquinolines in water.
Efficient synthesis of isoquinolines in water by a Pd-catalyzed tandem reaction of functionalized alkylnitriles with arylboronic acids
aCollege of Chemistry & Materials Engineering, Wenzhou University, Wenzhou 325035, China
E-mail: jiuxichen@wzu.edu.cn
bInstitute of Agricultural Resources and Regional Planning, Chinese Academy of Agricultural Sciences, Key Laboratory of Plant Nutrition and Fertilizer, Ministry of Agriculture, Beijing, China
Green Chem., 2017, Advance Article
DOI: 10.1039/C7GC00267J,
A palladium-catalyzed tandem reaction of 2-(cyanomethyl)benzonitriles or 2-(2-carbonylphenyl)acetonitriles with arylboronic acids in water has been developed for the first time. This reaction features good functional group tolerance and provides a new strategy for the synthesis of diverse isoquinolines under mild conditions. The use of water as the reaction medium makes the synthesis process environmentally benign. Preliminary mechanistic experiments indicate that the major reaction pathway involves carbopalladation of the C(sp3)–cyano group and subsequent intramolecular cyclization findings that were further supported by density functional theory (DFT) calculations.
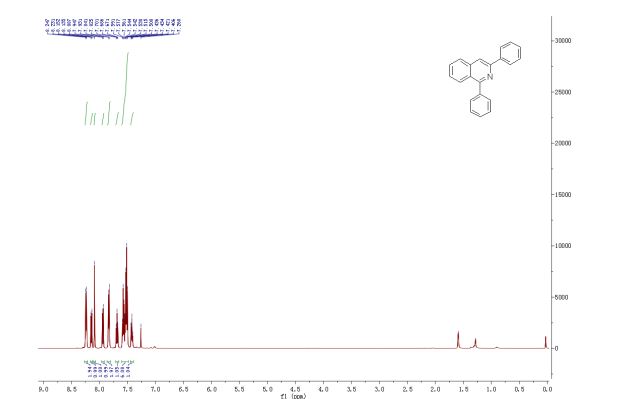
1,3-Diphenylisoquinoline (3a). Pale-yellow solid (103.5 mg, 92%),
mp 78-79 oC (lit.24, 73-74.5 oC). 24 J. D. Tovar and T. M. Swager, J. Org. Chem., 1999, 64, 6499
1H NMR (500 MHz, CDCl3) δ 8.25-8.23 (m, 2H), 8.15-8.14 (m, 1H), 8.09 (s, 1H), 7.95-7.93 (m, 1H), 7.84-7.83 (m, 2H), 7.70-7.67 (m, 1H), 7.59-7.50 (m, 6H), 7.44-7.40 (m, 1H);
13C NMR (125 MHz, CDCl3) δ 160.5, 150.3, 140.1, 139.8, 138.0, 130.4, 130.2, 128.8, 128.7, 128.6, 128.4, 127.7, 127.6, 127.2, 127.0, 126.0, 115.8.
//////// isoquinoline, pd-catalyzed, arylboronic acids


















![Graphical abstract: A two-step efficient preparation of a renewable dicarboxylic acid monomer 5,5′-[oxybis(methylene)]bis[2-furancarboxylic acid] from d-fructose and its application in polyester synthesis](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_v15y2tRSVduEsQJZWBXwGqKhEIbI4qULF4fct8H4sBtemhWWMU0MTh9HV2WpWqOixi5CPkoIIa0nYLt-ltSD9mfPfe4IOxygGe9mUL3IJiQjihM259fXZhkx6HcWbnPqQV3vTuR8F6g9SRtg9QsUMtjzuam2-yhHfHYBYsu35AYTL2NO7U8UnWX5lwRiP2zssfbFzNHhW7wKtGNBtR9yRwQw=s0-d)
















 :
: